Excited-state Properties Beyond the Excitation Energy from Orbital-Optimized Density Functional Calculations II: Absorption Spectra
Pith reviewed 2026-06-27 05:29 UTC · model grok-4.3
The pith
Orbital-optimized density functional calculations reproduce absorption spectra from high-level multireference methods across a molecular set.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
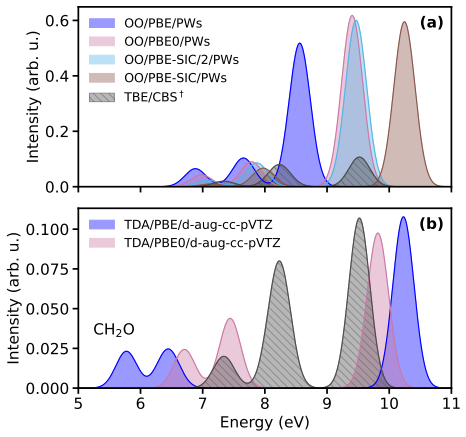
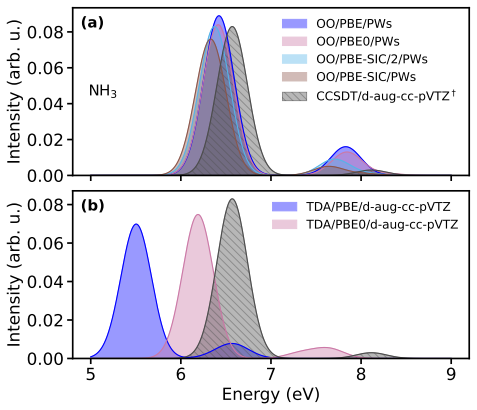
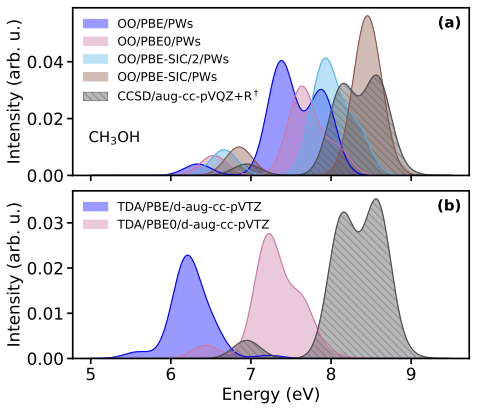
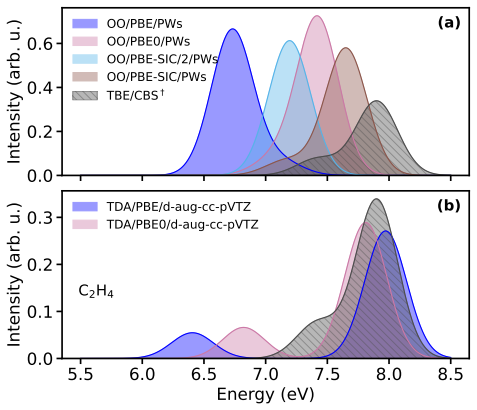
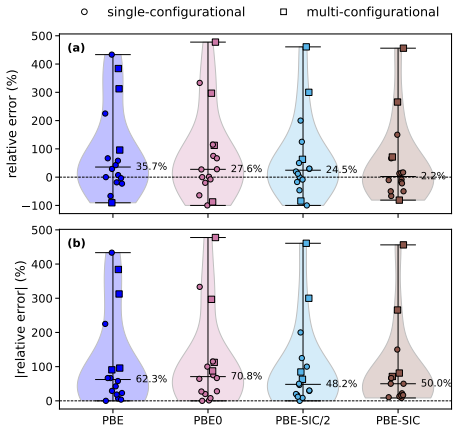
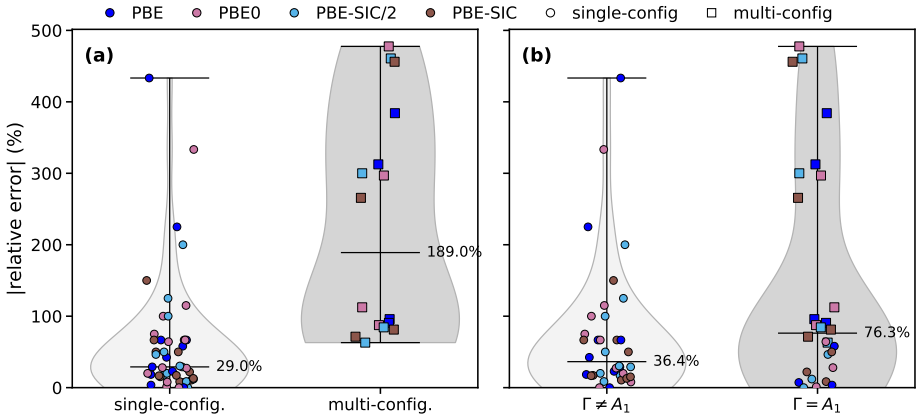
Orbital-optimized density functional calculations describe each excited state with its own variationally optimized orbitals and, after extension of Löwdin's formalism to the projector augmented-wave framework, yield oscillator strengths that allow reconstruction of absorption spectra. These spectra qualitatively match those from high-level multireference methods for the tested molecules even with a simple generalized-gradient approximation, with excellent intensity agreement for single-determinant states and larger discrepancies for multi-configurational states. Inclusion of exact exchange or self-interaction correction brings no systematic improvement, and nonzero overlaps between ground an
What carries the argument
Extension of Löwdin's formalism to the projector augmented-wave framework with plane-wave basis for oscillator strengths between nonorthogonal orbital-optimized states.
If this is right
- Absorption spectra become accessible from orbital-optimized density functional theory for sets of molecules without requiring advanced multireference methods.
- Peak intensities in the computed spectra match reference data well when the excited state is dominated by a single determinant.
- States with strong multi-configurational character produce substantial discrepancies in intensities that persist across different functionals.
- Adding exact exchange or explicit self-interaction correction does not systematically reduce errors in oscillator strengths.
- The magnitude of ground-excited state overlap does not predict the size of errors in the absorption peaks.
Where Pith is reading between the lines
- The method could enable spectrum calculations for larger molecules where multireference approaches remain computationally prohibitive.
- Focus on improving the underlying functional description for multi-configurational states may be needed to reduce remaining discrepancies.
- The absence of correlation with overlaps suggests the nonorthogonality treatment remains stable for the tested systems and basis sets.
Load-bearing premise
The extension of Löwdin's formalism to the projector augmented-wave framework with plane-wave basis accurately computes oscillator strengths without introducing uncontrolled errors from nonorthogonality handling or basis representation.
What would settle it
Direct numerical comparison of oscillator strengths computed for a strongly multi-configurational excited state against values from experiment or a higher-level multireference calculation would test whether the qualitative reproduction holds.
Figures

read the original abstract
Orbital-optimized density functional calculations describe each excited state using its own set of variationally optimized orbitals. While this state-specific optimization improves the description of excited states, it also results in nonorthogonal electronic states, which complicates the evaluation of transition properties. Existing benchmarks have primarily targeted the excitation energy, whereas estimates of oscillator strengths have largely been restricted to the first excited state only. In this study, L\"owdin's formalism for nonorthogonal determinants is extended to the projector augmented-wave framework. It is then used with a plane-wave basis set to compute the oscillator strengths of several valence and Rydberg excited states in a set of molecules. Orbital-optimized density functional calculations qualitatively reproduce absorption spectra from high-level multireference reference methods across the molecular set, even when using a simple generalized-gradient approximation. The agreement in peak intensities is excellent for states dominated by a single determinant, whereas substantial discrepancies arise for states with strong multi-configurational character. Analysis of the exchange--correlation functional shows that the inclusion of exact exchange and explicit self-interaction correction does not provide a systematic improvement in the accuracy of oscillator strengths. Moreover, no clear correlation emerges between the errors in the absorption peaks and the nonzero overlap between the ground state and the orbital-optimized excited states.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript extends Löwdin's formalism for transition matrix elements between nonorthogonal determinants to the projector augmented-wave (PAW) representation in a plane-wave basis. Orbital-optimized DFT calculations are then performed for valence and Rydberg excited states across a molecular test set, with oscillator strengths used to generate absorption spectra that are compared to high-level multireference reference data. The central claim is that even a simple GGA yields qualitative agreement with the reference spectra, with excellent intensity agreement for single-determinant states but larger discrepancies for multi-configurational states; no correlation is reported between ground-excited overlap and spectral errors, and inclusion of exact exchange or self-interaction correction does not systematically improve results.
Significance. If the Löwdin-PAW extension proves accurate, the work would show that state-specific orbital optimization in DFT can deliver reliable oscillator strengths beyond excitation energies, providing a parameter-free and computationally efficient route to absorption spectra that makes falsifiable predictions against external multireference benchmarks. This would be a useful advance for systems where multireference methods remain prohibitive.
major comments (3)
- [Theory section (Löwdin-PAW extension)] The extension of Löwdin's nonorthogonal-determinant formalism to the PAW plane-wave representation (described in the Theory section) is load-bearing for every reported oscillator strength and intensity. The manuscript supplies no numerical validation of this extension against analytically known oscillator strengths for nonorthogonal test cases, nor any convergence tests with respect to plane-wave cutoff or supercell size; without such checks, apparent agreement with multireference spectra could arise from uncontrolled basis or projector artifacts rather than the quality of the orbital-optimized densities.
- [Abstract and Results section] Abstract and Results section: the claim that peak intensities show 'excellent' agreement for single-determinant states and that the method 'qualitatively reproduces' the reference spectra is unsupported by any quantitative metric (e.g., mean absolute deviation, root-mean-square error, or integrated intensity ratios). Only qualitative language is used, which prevents a proportionate assessment of whether the Löwdin-PAW implementation is reliable enough to support the headline result.
- [Results section] Results section: the diagnostic that overlap magnitude does not correlate with error is presented, yet this test does not address possible systematic errors internal to the PAW implementation of the nonorthogonal transition formula (e.g., handling of nonorthogonality inside the projectors). A direct validation of the formalism itself remains necessary before the lack of correlation can be interpreted as evidence of robustness.
minor comments (2)
- [Abstract] The number of molecules, total states, and specific functionals employed should be stated explicitly in the abstract to give immediate context for the scope of the claimed qualitative agreement.
- [Figures] Figure captions and axis labels for the absorption spectra should include the precise definition of the broadening function and the units of the oscillator strength axis for reproducibility.
Simulated Author's Rebuttal
We thank the referee for their careful reading and constructive comments, which have identified important ways to strengthen the validation and presentation of our results. We address each major comment below and will incorporate the suggested improvements in a revised manuscript.
read point-by-point responses
-
Referee: [Theory section (Löwdin-PAW extension)] The extension of Löwdin's nonorthogonal-determinant formalism to the PAW plane-wave representation (described in the Theory section) is load-bearing for every reported oscillator strength and intensity. The manuscript supplies no numerical validation of this extension against analytically known oscillator strengths for nonorthogonal test cases, nor any convergence tests with respect to plane-wave cutoff or supercell size; without such checks, apparent agreement with multireference spectra could arise from uncontrolled basis or projector artifacts rather than the quality of the orbital-optimized densities.
Authors: We agree that explicit numerical validation of the Löwdin-PAW extension is necessary to confirm its accuracy independent of the molecular applications. In the revised manuscript we will add a dedicated subsection to the Theory section containing benchmark calculations against analytically known oscillator strengths for simple nonorthogonal determinant pairs, together with convergence tests with respect to plane-wave cutoff and supercell size. These additions will demonstrate that the reported spectral features are not dominated by basis-set or projector artifacts. revision: yes
-
Referee: [Abstract and Results section] Abstract and Results section: the claim that peak intensities show 'excellent' agreement for single-determinant states and that the method 'qualitatively reproduces' the reference spectra is unsupported by any quantitative metric (e.g., mean absolute deviation, root-mean-square error, or integrated intensity ratios). Only qualitative language is used, which prevents a proportionate assessment of whether the Löwdin-PAW implementation is reliable enough to support the headline result.
Authors: The referee correctly observes that the manuscript relies on qualitative descriptors without accompanying quantitative error metrics. We will revise the Abstract and Results sections to report mean absolute deviations, root-mean-square errors, and integrated intensity ratios for the oscillator strengths relative to the multireference reference data, separately for single-determinant and multi-configurational states. This will enable a more objective evaluation of the method's performance. revision: yes
-
Referee: [Results section] Results section: the diagnostic that overlap magnitude does not correlate with error is presented, yet this test does not address possible systematic errors internal to the PAW implementation of the nonorthogonal transition formula (e.g., handling of nonorthogonality inside the projectors). A direct validation of the formalism itself remains necessary before the lack of correlation can be interpreted as evidence of robustness.
Authors: We acknowledge that the overlap analysis alone does not exclude possible systematic errors arising from the PAW implementation of the nonorthogonal transition formula. The validation subsection added in response to the first comment will include targeted tests of nonorthogonality handling within the projectors and direct comparisons of transition matrix elements against known analytic results. These tests will provide the direct validation required to support the robustness of the reported lack of correlation. revision: yes
Circularity Check
No circularity; central results rest on external multireference benchmarks and established formalism extension
full rationale
The paper's core claim—that orbital-optimized DFT qualitatively reproduces absorption spectra—is validated by direct numerical comparison to independent high-level multireference reference methods on a molecular test set. The Löwdin nonorthogonal-determinant formalism is extended to the PAW plane-wave setting, but this extension is presented as a technical implementation step resting on prior established work rather than a self-referential fit or definition. No equations reduce a reported oscillator strength or spectral feature to a fitted parameter by construction, no uniqueness theorem is imported from the same authors to force the method, and no ansatz is smuggled via self-citation. Overlap diagnostics and functional comparisons are post-hoc analyses, not load-bearing inputs. The derivation chain therefore remains self-contained against external benchmarks.
Axiom & Free-Parameter Ledger
axioms (2)
- domain assumption Löwdin's formalism for nonorthogonal determinants can be accurately extended to the projector augmented-wave method without additional uncontrolled approximations.
- domain assumption Orbital-optimized DFT provides a valid description of excited states even with simple GGA functionals for the purpose of transition properties.
Reference graph
Works this paper leans on
-
[1]
Sarkar, Rudraditya and Boggio-Pasqua, Martial and Loos, Pierre-François and Jacquemin, Denis. Benchmarking TD-DFT and wave function methods for oscillator strengths and excited-state dipole moments. J. Chem. Theory Comput. doi:10.1021/acs.jctc.0c01228
-
[2]
Chutjian, A and Hall, R I and Trajmar, S. Electron-impact excitation of H2O and D2O at various scattering angles and impact energies in the energy-loss range 4.2–12 eV. J. Chem. Phys. doi:10.1063/1.431370
-
[3]
Chrayteh, Amara and Blondel, Aymeric and Loos, Pierre-François and Jacquemin, Denis. Mountaineering strategy to excited states: Highly accurate oscillator strengths and dipole moments of small molecules. J. Chem. Theory Comput. doi:10.1021/acs.jctc.0c01111
-
[4]
A theoretical determination of the electronic spectrum of formaldehyde
Merchán, Manuela and Roos, Björn O. A theoretical determination of the electronic spectrum of formaldehyde. Theoret. Chim. Acta. doi:10.1007/bf01125948
-
[5]
Excited states of the water molecule: analysis of the valence and Rydberg character
Rubio, Mercedes and Serrano-Andrés, Luis and Merchán, Manuela. Excited states of the water molecule: analysis of the valence and Rydberg character. J. Chem. Phys. doi:10.1063/1.2837827
-
[6]
Quantum Theory of Many-Particle Systems
Löwdin, Per-Olov. Quantum Theory of Many-Particle Systems. I. Physical Interpretations by Means of Density Matrices, Natural Spin-Orbitals, and Convergence Problems in the Method of Configurational Interaction. Phys. Rev. doi:10.1103/PhysRev.97.1474
-
[7]
Figari, Giuseppe and Magnasco, Valerio. On the evaluation of the cofactors occurring in the matrix elements between multiply-excited determinantal wavefunctions of non-orthogonal orbitals. Mol. Phys. doi:10.1080/00268978500101351
-
[8]
Generalized gradient approximation made simple
Perdew, J P and Burke, K and Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. doi:10.1103/PhysRevLett.77.3865
-
[9]
Generalized Gradient Approximation Made Simple [Phys
Perdew, John P and Burke, Kieron and Ernzerhof, Matthias. Generalized Gradient Approximation Made Simple [Phys. Rev. Lett. 77, 3865 (1996)]. Phys. Rev. Lett. doi:10.1103/PhysRevLett.78.1396
-
[11]
Projector augmented-wave method
Blöchl, P E. Projector augmented-wave method. Phys. Rev. B Condens. Matter. doi:10.1103/physrevb.50.17953
-
[12]
Electron affinities of the first-row atoms revisited
Kendall, Rick A and Dunning, Thom H and Harrison, Robert J. Electron affinities of the first-row atoms revisited. Systematic basis sets and wave functions. J. Chem. Phys. doi:10.1063/1.462569
-
[13]
A New Basis Set Exchange: An Open, Up-to-date Resource for the Molecular Sciences Community
Pritchard, Benjamin P and Altarawy, Doaa and Didier, Brett and Gibsom, Tara D and Windus, Theresa L. A New Basis Set Exchange: An Open, Up-to-date Resource for the Molecular Sciences Community. J. Chem. Inf. Model. doi:10.1021/acs.jcim.9b00725
-
[14]
Gaussian basis sets for use in correlated molecular calculations
Woon, David E and Dunning, Thom H. Gaussian basis sets for use in correlated molecular calculations. IV. Calculation of static electrical response properties. J. Chem. Phys. doi:10.1063/1.466439
-
[15]
Gaussian basis sets for use in correlated molecular calculations
Dunning, Thom H. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. doi:10.1063/1.456153
-
[16]
Software update: The ORCA program system—version 6.0
Neese, Frank. Software update: The ORCA program system—version 6.0. Wiley Interdiscip. Rev. Comput. Mol. Sci. doi:10.1002/wcms.70019
-
[17]
Neese, Frank. The ORCA program system. Wiley Interdiscip. Rev. Comput. Mol. Sci. doi:10.1002/wcms.81
-
[18]
Time-dependent density functional theory within the Tamm–Dancoff approximation
Hirata, So and Head-Gordon, Martin. Time-dependent density functional theory within the Tamm–Dancoff approximation. Chem. Phys. Lett. doi:10.1016/S0009-2614(99)01149-5
-
[19]
Real-space grid implementation of the projector augmented wave method
Mortensen, J J and Hansen, L B and Jacobsen, K W. Real-space grid implementation of the projector augmented wave method. Phys. Rev. B. doi:10.1103/PhysRevB.71.035109
-
[20]
a fer, Christian and J \'o nsson, Elvar \
Mortensen, Jens J rgen and Larsen, Ask Hjorth and Kuisma, Mikael and Ivanov, Aleksei V and Taghizadeh, Alireza and Peterson, Andrew and Haldar, Anubhab and Dohn, Asmus Ougaard and Sch \"a fer, Christian and J \'o nsson, Elvar \"O rn and Hermes, Eric D and Nilsson, Fredrik Andreas and Kastlunger, Georg and Levi, Gianluca and J \'o nsson, Hannes and H \"a k...
-
[21]
Self-consistent field calculations of excited states using the maximum overlap method (MOM)
Gilbert, Andrew T B and Besley, Nicholas A and Gill, Peter M W. Self-consistent field calculations of excited states using the maximum overlap method (MOM). J. Phys. Chem. A. doi:10.1021/jp801738f
-
[22]
Hait, Diptarka and Head-Gordon, Martin. Highly accurate prediction of core spectra of molecules at density functional theory cost: Attaining sub-electronvolt error from a restricted open-shell Kohn-Sham approach. J. Phys. Chem. Lett. doi:10.1021/acs.jpclett.9b03661
-
[23]
Reliable transition properties from excited-state mean-field calculations
Bourne Worster, Susannah and Feighan, Oliver and Manby, Frederick R. Reliable transition properties from excited-state mean-field calculations. J. Chem. Phys. doi:10.1063/5.0041233
-
[24]
Highly accurate and robust constraint-based orbital-optimized core excitations
Lemke, Yannick and Kussmann, Jörg and Ochsenfeld, Christian. Highly accurate and robust constraint-based orbital-optimized core excitations. J. Phys. Chem. A. doi:10.1021/acs.jpca.4c04139
-
[25]
Optimization of Wave Function and Geometry in the Finite Basis Hartree-Fock Method , volume =
Martin Head-Gordon and John A Pople , issue =. Optimization of Wave Function and Geometry in the Finite Basis Hartree-Fock Method , volume =. J. Phys. Chem , pages =
-
[26]
Hait, Diptarka and Haugen, Eric A and Yang, Zheyue and Oosterbaan, Katherine J and Leone, Stephen R and Head-Gordon, Martin. Accurate prediction of core-level spectra of radicals at density functional theory cost via square gradient minimization and recoupling of mixed configurations. J. Chem. Phys. doi:10.1063/5.0018833
-
[27]
Selenius, Elli and Sigurdarson, Alec Elías and Schmerwitz, Yorick L A and Levi, Gianluca. Orbital-optimized versus time-dependent density functional calculations of intramolecular charge transfer excited states. J. Chem. Theory Comput. doi:10.1021/acs.jctc.3c01319
-
[28]
Faraday Discussions , volume=
Variational calculations of excited states via direct optimization of the orbitals in DFT , author=. Faraday Discussions , volume=. 2020 , publisher=
2020
-
[29]
Ivanov, Aleksei V. and Levi, Gianluca and J. J. Chem. Theory Comput. , month =. doi:10.1021/acs.jctc.1c00157 , issn =
-
[30]
Sigurdarson, Alec E and Schmerwitz, Yorick L A and Tveiten, Dagrún K V and Levi, Gianluca and Jónsson, Hannes. Orbital-optimized density functional calculations of molecular Rydberg excited states with real space grid representation and self-interaction correction. J. Chem. Phys. doi:10.1063/5.0179271
-
[31]
Variational density functional calculations of excited states via direct optimization
Levi, Gianluca and Ivanov, Aleksei V and Jónsson, Hannes. Variational density functional calculations of excited states via direct optimization. J. Chem. Theory Comput. doi:10.1021/acs.jctc.0c00597
-
[32]
Role of Rydberg states in the photochemical dynamics of ethylene
Mori, Toshifumi and Glover, William J and Schuurman, Michael S and Martinez, Todd J. Role of Rydberg states in the photochemical dynamics of ethylene. J. Phys. Chem. A. doi:10.1021/jp2097185
-
[33]
Rydberg states: sensitive probes of molecular structure
Kuthirummal, Narayanan and Weber, Peter M. Rydberg states: sensitive probes of molecular structure. Chem. Phys. Lett. doi:10.1016/j.cplett.2003.08.002
-
[34]
Molecules in high Rydberg states
Merkt, F. Molecules in high Rydberg states. Annu. Rev. Phys. Chem. doi:10.1146/annurev.physchem.48.1.675
-
[35]
The role of Rydberg states in spectroscopy and photochemistry: Low and high Rydberg states
Sándorfy, C. The role of Rydberg states in spectroscopy and photochemistry: Low and high Rydberg states. doi:10.1007/0-306-46938-3
-
[36]
Jochim, Bethany and Siemering, R and Zohrabi, M and Voznyuk, O and Mahowald, J B and Schmitz, D G and Betsch, K J and Berry, Ben and Severt, T and Kling, Nora G and Burwitz, T G and Carnes, K D and Kling, M F and Ben-Itzhak, I and Wells, E and de Vivie-Riedle, R. The importance of Rydberg orbitals in dissociative ionization of small hydrocarbon molecules ...
-
[37]
van Harrevelt, Rob and van Hemert, Marc C. Photodissociation of water. I. Electronic structure calculations for the excited states. J. Chem. Phys. doi:10.1063/1.481153
-
[38]
Applications of molecular Rydberg states in chemical dynamics and spectroscopy
Softley, T P. Applications of molecular Rydberg states in chemical dynamics and spectroscopy. Int. Rev. Phys. Chem. doi:10.1080/01442350310001652940
-
[39]
Feller, David and Peterson, Kirk A and Davidson, Ernest R. A systematic approach to vertically excited states of ethylene using configuration interaction and coupled cluster techniques. J. Chem. Phys. doi:10.1063/1.4894482
-
[40]
A mountaineering strategy to excited states: Highly accurate reference energies and benchmarks
Loos, Pierre-François and Scemama, Anthony and Blondel, Aymeric and Garniron, Yann and Caffarel, Michel and Jacquemin, Denis. A mountaineering strategy to excited states: Highly accurate reference energies and benchmarks. J. Chem. Theory Comput. doi:10.1021/acs.jctc.8b00406
-
[41]
Applications of time-dependent and time-independent density functional theory to Rydberg transitions
Seidu, Issaka and Krykunov, Mykhaylo and Ziegler, Tom. Applications of time-dependent and time-independent density functional theory to Rydberg transitions. J. Phys. Chem. A. doi:10.1021/jp5082802
-
[42]
Ammonia: the prototypical lone pair molecule
Bartlett, Rodney J and Del Bene, Janet E and Perera, S Ajith and Mattie, Reneépeloquin. Ammonia: the prototypical lone pair molecule. Theochem. doi:10.1016/s0166-1280(97)90277-3
-
[43]
Journal of Chemical Theory and Computation , pages =
Levi, Gianluca and Kroesbergen, Max and Thirion, Louis and Schmerwitz, Yorick L A and Elvar, O J and Bilous, Pavlo and Hansmann, Philipp and Hannes, J , doi =. Journal of Chemical Theory and Computation , pages =
-
[44]
Journal of Chemical Theory and Computation , pages =
Schmerwitz, Yorick L A and Selenius, Elli and Levi, Gianluca , doi =. Journal of Chemical Theory and Computation , pages =
-
[45]
Schmerwitz, Yorick L. A. and Levi, Gianluca and J. J. Chem. Theory Comput. , number =. doi:10.1021/acs.jctc.3c00178 , eprint =
-
[46]
General-model-space state-universal coupled-cluster method: excitation energies of water
Li, Xiangzhu and Paldus, Josef. General-model-space state-universal coupled-cluster method: excitation energies of water. Mol. Phys. doi:10.1080/00268970500416145
-
[47]
Ciofini, Ilaria and Adamo, Carlo. Accurate evaluation of valence and low-lying Rydberg states with standard time-dependent density functional theory. J. Phys. Chem. A. doi:10.1021/jp0722152
-
[48]
Universal Gaussian basis sets for an optimum representation of Rydberg and continuum wavefunctions
Kaufmann, K and Baumeister, W and Jungen, M. Universal Gaussian basis sets for an optimum representation of Rydberg and continuum wavefunctions. J. Phys. B At. Mol. Opt. Phys. doi:10.1088/0953-4075/22/14/007
-
[49]
Simultaneous calculation of Rydberg and valence excited states of formaldehyde
Müller, Thomas and Lischka, Hans. Simultaneous calculation of Rydberg and valence excited states of formaldehyde. Theor. Chem. Acc. doi:10.1007/s002140100286
-
[50]
Zobel, J Patrick and Nogueira, Juan J and González, Leticia. The IPEA dilemma in CASPT2. Chem. Sci. doi:10.1039/c6sc03759c
-
[51]
Automated active space selection with dipole moments
Kaufold, Benjamin W and Chintala, Nithin and Pandeya, Pratima and Dong, Sijia S. Automated active space selection with dipole moments. J. Chem. Theory Comput. doi:10.1021/acs.jctc.2c01128
-
[52]
Veryazov, Valera and Malmqvist, Per ke and Roos, Bj \"o rn O. How to select active space for multiconfigurational quantum chemistry?: Selection of Active Space for Multiconfigurational Methods. Int. J. Quantum Chem. doi:10.1002/qua.23068
-
[53]
Theoretical study of the absorption and emission spectra of indole in the gas phase and in a solvent
Serrano-Andrés, Luis and Roos, Björn O. Theoretical study of the absorption and emission spectra of indole in the gas phase and in a solvent. J. Am. Chem. Soc. doi:10.1021/ja952035i
-
[54]
Theoretical study of the electronic spectrum of imidazole
Serrano-Andrés, Luis and Fülscher, Markus P and Roos, Björn O and Merchán, Manuela. Theoretical study of the electronic spectrum of imidazole. J. Phys. Chem. doi:10.1021/jp952809h
-
[55]
Excited state dipole moments from SCF: a benchmark
Paetow, Lukas and Neugebauer, Johannes. Excited state dipole moments from SCF: a benchmark. Phys. Chem. Chem. Phys. 2025 , pages =. doi:10.1039/d5cp01695a
-
[56]
Rationale for mixing exact exchange with density functional approximations
Perdew, John P and Ernzerhof, Matthias and Burke, Kieron. Rationale for mixing exact exchange with density functional approximations. J. Chem. Phys. doi:10.1063/1.472933
-
[57]
Self-interaction correction to density-functional approximations for many-electron systems
Perdew, J P and Zunger, Alex. Self-interaction correction to density-functional approximations for many-electron systems. Phys. Rev. B Condens. Matter. doi:10.1103/physrevb.23.5048
-
[58]
Melander, Marko and J \'o nsson, Elvar \"O and Mortensen, Jens J and Vegge, Tejs and Garc \' a Lastra, Juan Maria. Implementation of constrained DFT for computing charge transfer rates within the projector augmented wave method. J. Chem. Theory Comput. doi:10.1021/acs.jctc.6b00815
-
[59]
Systematic optimization of long-range corrected hybrid density functionals
Chai, Jeng-Da and Head-Gordon, Martin. Systematic optimization of long-range corrected hybrid density functionals. J. Chem. Phys. doi:10.1063/1.2834918
-
[60]
Absolute photoabsorption cross-sections of methanol for terrestrial and astrophysical relevance
Lange, Emanuele and Lozano, Ana Isabel and Jones, Nykola C and Hoffmann, Søren Vrønning and Kumar, Sarvesh and Śmiałek, Małgorzata A and Duflot, Denis and Brunger, Michael J and Limão-Vieira, Paulo. Absolute photoabsorption cross-sections of methanol for terrestrial and astrophysical relevance. J. Phys. Chem. A. doi:10.1021/acs.jpca.0c06615
-
[61]
On the calculation of multiplet energies by the hartree-fock-slater method
Ziegler, Tom and Rauk, Arvi and Baerends, Evert J. On the calculation of multiplet energies by the hartree-fock-slater method. Theoret. Chim. Acta. doi:10.1007/bf00551551
-
[62]
Yang, Ke and Peverati, Roberto and Truhlar, Donald G and Valero, Rosendo. Density functional study of multiplicity-changing valence and Rydberg excitations of p-block elements: delta self-consistent field, collinear spin-flip time-dependent density functional theory (DFT), and conventional time-dependent DFT. J. Chem. Phys. doi:10.1063/1.3607312
-
[63]
Recent Advances In Density Functional Methods: (Part I) , pages=
Time-dependent density functional response theory for molecules , author=. Recent Advances In Density Functional Methods: (Part I) , pages=. 1995 , publisher=
1995
-
[64]
Physical review , volume=
Inhomogeneous electron gas , author=. Physical review , volume=. 1964 , publisher=
1964
-
[65]
K U , doi =
Runge, Erich and Gross, E. K U , doi =. Phys. Rev. Lett. , number =
-
[66]
Rydberg energies using excited state density functional theory , author=. J. Chem. Phys. , volume=. 2008 , publisher=
2008
-
[67]
and Benfield, Peter and Helgaker, Trygve and Tozer, David J
Peach, Michael J.G. and Benfield, Peter and Helgaker, Trygve and Tozer, David J. , doi =. Journal of Chemical Physics , number =
-
[68]
and Jamorski, Christine and Casida, Kim C
Casida, Mark E. and Jamorski, Christine and Casida, Kim C. and Salahub, Dennis R. , doi =. Journal of Chemical Physics , number =
-
[69]
Journal of the American Chemical Society , number =
Shu, Yinan and Truhlar, Donald G , doi =. Journal of the American Chemical Society , number =
-
[70]
Buenker, Robert J and Peyerimhoff, Sigrid D. Mixed valence—Rydberg states. Chem. Phys. Lett. doi:10.1016/0009-2614(75)80271-5
-
[71]
Toffoli, Daniele and Quarin, Matteo and Fronzoni, Giovanna and Stener, Mauro. Accurate vertical excitation energies of BODIPY/Aza-BODIPY derivatives from excited-state mean-field calculations. J. Phys. Chem. A. doi:10.1021/acs.jpca.2c04473
-
[72]
Vandaele, Eva and Mališ, Momir and Luber, Sandra. The photodissociation of solvated cyclopropanone and its hydrate explored via non-adiabatic molecular dynamics using SCF. Phys. Chem. Chem. Phys. doi:10.1039/d1cp05187c
-
[73]
Borges, Jr, Itamar. Configuration interaction oscillator strengths of the H2O molecule: Transitions from the ground to the B ^1 A _1 , C ^1 1B _1 , D ^1 1A _1 , and 11B2 excited states. Chem. Phys. doi:10.1016/j.chemphys.2006.07.007
-
[74]
Ab initio study of valence and Rydberg states of CH3Br
Escure, Christelle and Leininger, Thierry and Lepetit, Bruno. Ab initio study of valence and Rydberg states of CH3Br. J. Chem. Phys. doi:10.1063/1.3152865
-
[75]
Zheng, Lianjun and Polizzi, Nicholas F and Dave, Adarsh R and Migliore, Agostino and Beratan, David N. Where is the electronic oscillator strength? Mapping oscillator strength across molecular absorption spectra. J. Phys. Chem. A. doi:10.1021/acs.jpca.6b00692
-
[76]
Reisler, Hanna and Krylov, Anna I. Interacting Rydberg and valence states in radicals and molecules: experimental and theoretical studies. Int. Rev. Phys. Chem. doi:10.1080/01442350902989170
-
[77]
doi:10.1021/ct500727c , issn =
Journal of Chemical Theory and Computation , number =. doi:10.1021/ct500727c , issn =
-
[78]
Lacombe, Lionel and Maitra, Neepa T. , doi =. npj Computational Materials , number =. 2023 , pages =. arXiv , arxivId =:2302.11366 , issn =
arXiv 2023
-
[79]
An ab initio exciton model including charge-transfer excited states
Li, Xin and Parrish, Robert M and Liu, Fang and Kokkila Schumacher, Sara I L and Martínez, Todd J. An ab initio exciton model including charge-transfer excited states. J. Chem. Theory Comput. doi:10.1021/acs.jctc.7b00171
-
[80]
On the determination of excitation energies using density functional theory
Tozer, David J and Handy, Nicholas C. On the determination of excitation energies using density functional theory. Phys. Chem. Chem. Phys. doi:10.1039/a910321j
-
[81]
Palmer, Michael H and Gordon, Agnieszka J. The electronic states of isothiazole studied by VUV absorption spectroscopy and ab initio configuration interaction methods. Chem. Phys. doi:10.1016/j.chemphys.2007.09.044
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.