Arch\^e, an orbital-free molecular dynamics code for fast production of equations of state
Pith reviewed 2026-06-27 05:28 UTC · model grok-4.3
The pith
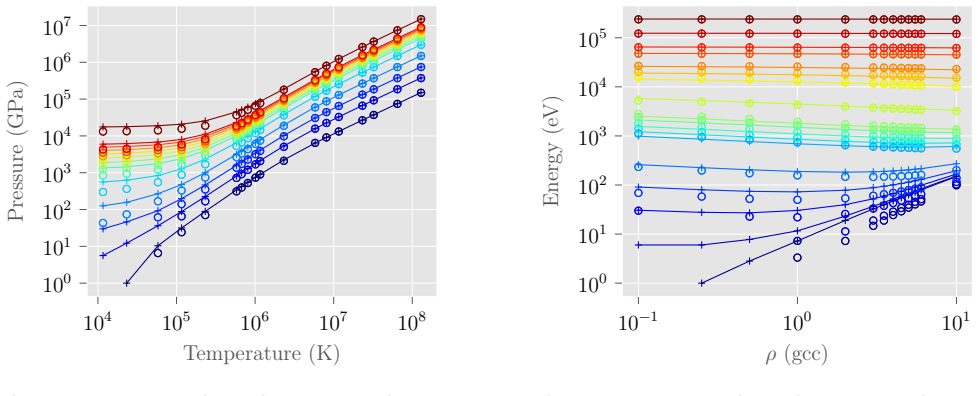
Archê produces aluminum equations of state matching Abinit results using an orbital-free approach after applying an average-atom pseudopotential correction.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
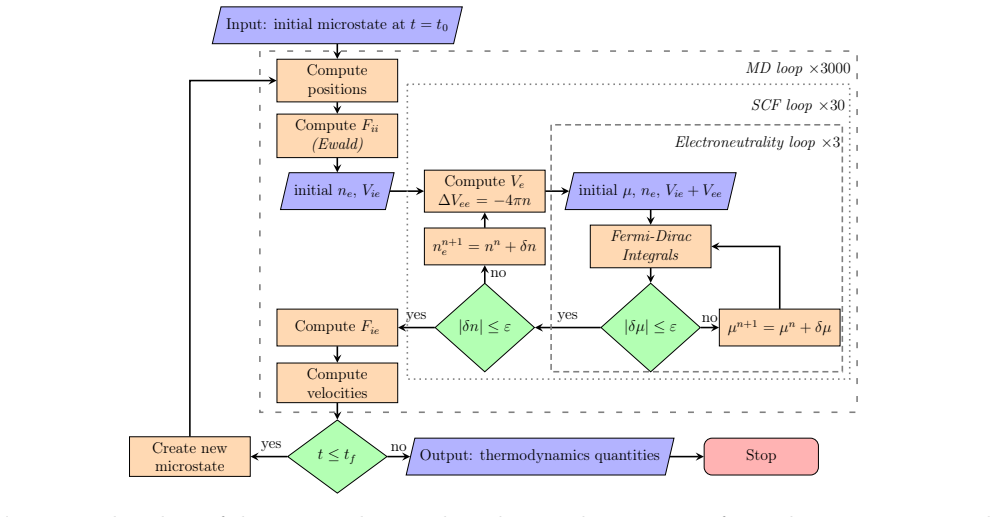
Archê implements orbital-free molecular dynamics with a self-consistent field solver for the electronic density, accelerated by timestep-based density initialization and free-energy-minimizing mixing. When a pseudopotential correction derived from an average-atom model is included, the resulting internal energies for aluminum match those from Kohn-Sham calculations in Abinit. The method scales linearly with system size and runs an order of magnitude faster on a single GPU than on 256 CPUs.
What carries the argument
The self-consistent field computation of electronic density in orbital-free MD, initialized from prior timesteps and accelerated by approximate free-energy density mixing, together with the average-atom pseudopotential correction.
If this is right
- Equations of state for plasmas can be generated with linear scaling in number of atoms and grid points.
- SCF convergence is accelerated by up to six times compared to standard methods.
- Execution time decreases with increasing temperature, unlike orbital-based simulations.
- A single GPU achieves an order-of-magnitude speedup over 256 CPUs.
- Accurate internal energies require the added pseudopotential correction for agreement with full DFT.
Where Pith is reading between the lines
- The approach may enable simulations of larger systems or longer times in plasma physics that were previously computationally prohibitive.
- Similar corrections might be needed or adaptable for other materials beyond aluminum.
- The temperature dependence could lead to more efficient high-temperature plasma modeling.
Load-bearing premise
The orbital-free density functional approximation combined with the average-atom pseudopotential correction is assumed to be adequate for accurate internal energies across the relevant plasma conditions without additional material-specific parameters.
What would settle it
A direct comparison of internal energy values from Archê and Abinit for aluminum at a density and temperature where the difference exceeds the agreement shown in the paper's validation plots.
Figures







read the original abstract
We present Arch\^e, an orbital-free molecular dynamics (OFMD) code designed to produce equations of state (EOS) in the plasma state. Unlike in other OFMD codes, Arch\^e uses a self-consistent field (SCF) approach to compute the electronic density. This allows us to implement two algorithms that accelerate SCF convergence by a factor of up to six. First, the density is initialized using the results from the previous MD timestep to define a one-center profile, which is then applied to the new nuclei positions. Second, the initial and final densities are mixed at each SCF iteration in a proportion that minimizes an approximate free energy. We validate the code by comparing the calculated aluminum EOS to results obtained with the Kohn-Sham density functional theory software Abinit. Achieving agreement in the internal energy requires adding a correction related to the norm-conserving pseudopotential derived from an average-atom model. Performance is compared across CPU and GPU architectures, demonstrating an order-of-magnitude speedup for a single GPU compared to 256 CPUs. Arch\^e exhibits an overall linear computational complexity with respect to the number of atoms, as well as the number of real and reciprocal grid points. Execution time is weakly dependent on density; however, interestingly, it decreases as temperature increases -- in contrast to simulations based on Kohn-Sham orbitals.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript presents Archê, an orbital-free molecular dynamics (OFMD) code for producing equations of state in the plasma regime. It employs a self-consistent field (SCF) approach to the electronic density (in contrast to other OFMD implementations) and introduces two acceleration techniques: (i) initializing the density at each MD step from a one-center profile derived from the previous timestep, and (ii) mixing initial and final densities at each SCF iteration to minimize an approximate free energy. These yield up to 6× faster SCF convergence. Validation is performed on aluminum EOS internal energies against the Kohn-Sham DFT code Abinit; agreement is achieved only after adding a norm-conserving pseudopotential correction taken from an average-atom model. The code is shown to exhibit linear scaling with atom number and grid size, an order-of-magnitude GPU speedup relative to 256 CPUs, and execution time that decreases with temperature.
Significance. If the accuracy claim holds after the correction, Archê would offer a practical route to high-throughput plasma EOS tables at conditions where Kohn-Sham orbital methods become prohibitive. The explicit cross-code validation against Abinit, the linear complexity, and the GPU performance data are concrete strengths. However, the necessity of an average-atom-derived correction for internal-energy agreement indicates that the base orbital-free SCF treatment is not yet sufficient on its own, which tempers the immediate generality of the method.
major comments (2)
- [Validation / Abstract] Validation results (abstract and corresponding section): agreement with Abinit internal energies is stated to require the addition of a norm-conserving pseudopotential correction derived from an average-atom model. The manuscript should quantify the size of the discrepancy without this correction, identify its physical origin (orbital-free kinetic-energy functional, pseudopotential, or SCF implementation), and demonstrate whether the same correction remains accurate when applied to full multi-center MD trajectories or to other materials without re-derivation.
- [Performance benchmarks] Performance claims (abstract and benchmarks section): the reported factors (up to 6× SCF acceleration and order-of-magnitude GPU vs. 256-CPU speedup) are presented without accompanying statistical measures (standard deviations, number of independent runs, or dependence on system size). Because MD timings can fluctuate, these quantitative claims need error bars or tabulated raw data to be load-bearing for the central performance assertion.
minor comments (2)
- [Abstract] Abstract: the statement that execution time 'decreases as temperature increases' is interesting but would benefit from a brief physical explanation or reference to the underlying algorithmic scaling.
- [Methods] Notation: the manuscript should explicitly define the approximate free-energy functional used in the mixing step and state whether it is evaluated on the same grid as the full SCF calculation.
Simulated Author's Rebuttal
We thank the referee for their careful reading and constructive comments. Below we respond point by point to the major comments.
read point-by-point responses
-
Referee: [Validation / Abstract] Validation results (abstract and corresponding section): agreement with Abinit internal energies is stated to require the addition of a norm-conserving pseudopotential correction derived from an average-atom model. The manuscript should quantify the size of the discrepancy without this correction, identify its physical origin (orbital-free kinetic-energy functional, pseudopotential, or SCF implementation), and demonstrate whether the same correction remains accurate when applied to full multi-center MD trajectories or to other materials without re-derivation.
Authors: We agree that explicit quantification strengthens the validation section. The revised manuscript will add a table reporting the internal-energy discrepancy (without the correction) for the aluminum cases examined; the difference is several eV/atom and is localized to the core region. We identify the origin as the norm-conserving pseudopotential, because the orbital-free kinetic-energy functional and SCF solver are standard and produce consistent densities; the average-atom model supplies the missing core correction. The existing validation already uses configurations taken from multi-center MD trajectories, so the correction is applied in that setting. For other materials we have not performed the test and will therefore add an explicit statement that the correction is material-specific and would require re-derivation; this is noted as a current limitation rather than a claim of immediate generality. revision: partial
-
Referee: [Performance benchmarks] Performance claims (abstract and benchmarks section): the reported factors (up to 6× SCF acceleration and order-of-magnitude GPU vs. 256-CPU speedup) are presented without accompanying statistical measures (standard deviations, number of independent runs, or dependence on system size). Because MD timings can fluctuate, these quantitative claims need error bars or tabulated raw data to be load-bearing for the central performance assertion.
Authors: We accept the point. The revised manuscript will state that each reported timing is the average of five independent runs on the same hardware, will include the corresponding standard deviations, and will tabulate raw wall-clock times versus atom number and grid size so that the linear-scaling and GPU/CPU ratios can be inspected directly. revision: yes
Circularity Check
No significant circularity; validation against independent Abinit and performance benchmarks are self-contained.
full rationale
The paper introduces Archê as an OFMD code using SCF with two acceleration algorithms (prior-timestep density initialization and free-energy-minimizing mixing). The central accuracy claim for aluminum EOS internal energy is achieved only after adding an external average-atom-derived pseudopotential correction, but this addition is presented as a required post-processing step rather than a fitted parameter renamed as a prediction. No equations reduce claimed results to quantities defined or fitted within the paper itself. Performance scaling (linear complexity, GPU speedup) is demonstrated via direct benchmarks against CPU runs and Abinit, with no self-citation load-bearing the core claims and no self-definitional loops. The derivation chain for the SCF solvers and complexity analysis remains independent of the target EOS data.
Axiom & Free-Parameter Ledger
axioms (1)
- domain assumption Orbital-free density functional theory approximations remain valid for the plasma regime when supplemented by the average-atom pseudopotential correction
Reference graph
Works this paper leans on
-
[1]
Burke, Perspective on density functional theory., The Journal of chemical physics 136 (2012) 150901
K. Burke, Perspective on density functional theory., The Journal of chemical physics 136 (2012) 150901
2012
-
[2]
Kittel, Introduction to Solid State Physics, 8th Edition, Wiley, 2004
C. Kittel, Introduction to Solid State Physics, 8th Edition, Wiley, 2004
2004
-
[3]
Kresse, J
G. Kresse, J. Furthmüller, Efficiency of ab- initio total energy calculations for metals and semiconductors using a plane-wave basis set, Computational Materials Science 6 (1996) 15–50
1996
-
[4]
Blanchet, M
A. Blanchet, M. Torrent, J. Clérouin, Requirements for very high temperature Kohn–Sham DFT simulations and how to bypass them, Physics of Plasmas 27 (2020) 122706
2020
-
[5]
S. Yin, Y. Zuo, A. Abu-Odeh, H. Zheng, X.-G. Li, J. Ding, S. P. Ong, M. Asta, R. O. Ritchie, Atomistic simulations of dislo- cation mobility in refractory high-entropy al- loys and the effect of chemical short-range or- der, Nature Communications 12 (2021) 4873
2021
-
[6]
B. A. Remington, R. P. Drake, D. D. Ryutov, Experimental astrophysics with high power lasers and Z pinches, Reviews of Modern Physics 78 (2006) 755
2006
-
[7]
J. D. Lindl, Development of the indirect- drive approach to inertial confinement fusion and the target physics basis for ignition and gain, Physics of Plasmas 2 (1995) 3933
1995
-
[8]
H. Abu-Shawareb, R. Acree, P. Adams, J. Adams, B. Addis, R. Aden, P. Adrian, B. B. Afeyan, M. Aggleton, L. Aghaian, A. Aguirre, D. Aikens, J. Akre, F. Albert, M. Albrecht, B. J. Albright, J. Albrit- ton, J. Alcala, C. Alday, D. A. Alessi, N. Alexander, J. Alfonso, N. Alfonso, E. Al- ger, S. J. Ali, Z. A. Ali, A. Allen, W. E. Alley, P. Amala, P. A. Amendt,...
-
[9]
H. Abu-Shawareb, R. Acree, P. Adams, J. Adams, B. Addis, R. Aden, P. Adrian, B. B. Afeyan, M. Aggleton, L. Aghaian, A. Aguirre, D. Aikens, J. Akre, F. Albert, M. Albrecht, B. J. Albright, J. Albrit- ton, J. Alcala, C. Alday, D. A. Alessi, N. Alexander, J. Alfonso, N. Alfonso, E. Alger, S. J. Ali, Z. A. Ali, W. E. Al- ley, P. Amala, P. A. Amendt, P. Amick,...
-
[10]
Bonitz, T
M. Bonitz, T. Dornheim, Z. A. Moldabekov, S. Zhang, P. Hamann, H. Kählert, A. Fili- nov, K. Ramakrishna, J. Vorberger, Ab ini- tio simulation of warm dense matter, Physics of Plasmas 27 (2020) 042710
2020
-
[11]
Suryanarayana, P
P. Suryanarayana, P. P. Pratapa, A. Sharma, J. E. Pask, SQDFT: Spectral quadrature method for large-scale parallel o(N) Kohn- Sham calculations at high temperature, Comput. Phys. Commun. 224 (2018) 288– 298
2018
-
[12]
Zhang, H
S. Zhang, H. Wang, W. Kang, P. Zhang, X. T. He, Extended application of Kohn- Sham first-principles molecular dynamics method with plane wave approximation at high energy - from cold materials to hot dense plasmas, Physics of Plasmas 23 (2016) 042707
2016
-
[13]
A. Blanchet, J. Clérouin, M. Torrent, F. Soubiran, Extended first-principles molec- ular dynamics model for high temperature simulations in the Abinit code: Application to warm dense aluminum, Comput. Phys. Commun. 271 (2022) 108215.doi:https: //doi.org/10.1016/j.cpc.2021.108215. URLhttps://www.sciencedirect. com/science/article/pii/ S0010465521003271 28
-
[14]
R. Baer, D. Neuhauser, E. Rabani, Self- averaging stochastic Kohn-Sham density- functional theory., Physical Review Letters 111 (2013) 106402
2013
-
[15]
A. J. White, L. A. Collins, Fast and univer- sal Kohn-Sham density functional theory al- gorithm for warm dense matter to hot dense plasma., Physical Review Letters 125 (2020) 055002
2020
-
[16]
Q. Liu, M. Chen, Plane-wave-based stochastic-deterministic density func- tional theory for extended systems, Physical Review B 106 (2022) 125132
2022
-
[17]
Sharma, L
V. Sharma, L. A. Collins, A. J. White, Stochastic and mixed density functional the- ory within the projector augmented wave for- malism for simulation of warm dense matter., Physical review E 108 (2023) L023201
2023
-
[18]
Blanchet, From the surface to the core of stars: towards a unified modeling from condensed matter to plasmas, Ph.D
A. Blanchet, From the surface to the core of stars: towards a unified modeling from condensed matter to plasmas, Ph.D. thesis (2022)
2022
-
[19]
W. Mi, K. Luo, S. B. Trickey, M. Pavanello, Orbital-free density functional theory: An attractive electronic structure method for large-scale first-principles simulations, Chemical Reviews 123 (21) (2023) 12039– 12104, pMID: 37870767.arXiv:https:// doi.org/10.1021/acs.chemrev.2c00758, doi:10.1021/acs.chemrev.2c00758. URLhttps://doi.org/10.1021/acs. chemre...
-
[20]
V. V. Karasiev, K. P. Hilleke, S. B. Trickey, Free-energy orbital-free den- sity functional theory: recent develop- ments, perspective, and outlook, Elec- tronic Structure 7 (1) (2025) 013001. doi:10.1088/2516-1075/adadd4. URLhttps://doi.org/10.1088/ 2516-1075/adadd4
-
[21]
R. P. Feynman, N. Metropolis, E. Teller, Equations of state of elements based on the generalized Fermi-Thomas theory, Phys. Rev. 75 (1949) 1561
1949
-
[22]
K. Luo, V. V. Karasiev, S. B. Trickey, To- wards accurate orbital-free simulations: A generalized gradient approximation for the noninteractingfreeenergydensityfunctional, Phys. Rev. B 101 (2020) 075116
2020
-
[23]
V. Rios-Vargas, X. Shao, S. B. Trickey, M. Pavanello, Effective Wang-Teter kernels for improved orbital-free den- sity functional theory simulations, Phys. Rev. B 110 (2024) 085129. doi:10.1103/PhysRevB.110.085129. URLhttps://link.aps.org/doi/10. 1103/PhysRevB.110.085129
-
[24]
A. Bhattacharjee, S. Jana, P. Samal, First step toward a parameter-free, nonlocal kinetic energy density functional for semi- conductors and simple metals, The Journal of Chemical Physics 160 (22) (2024) 224110. arXiv:https://pubs.aip.org/aip/jcp/ article-pdf/doi/10.1063/5.0204957/ 19998017/224110_1_5.0204957.pdf, doi:10.1063/5.0204957. URLhttps://doi.org...
-
[25]
J. P. Perdew, K. Schmidt, Jacob’s ladder of density functional approxi- mations for the exchange-correlation energy, AIP Conference Proceedings 577 (1) (2001) 1–20.arXiv:https: //pubs.aip.org/aip/acp/article-pdf/ 577/1/1/12108089/1_1_online.pdf, doi:10.1063/1.1390175. URLhttps://doi.org/10.1063/1. 1390175
-
[26]
D. I. Mihaylov, S. Hu, V. V. Karasiev, Dragon: A multi-GPU orbital-free den- sity functional theory molecular dynam- ics simulation package for modeling of warm dense matter, Comput. Phys. Com- mun. 294 (2024) 108931.doi:https: //doi.org/10.1016/j.cpc.2023.108931. URLhttps://www.sciencedirect. 29 com/science/article/pii/ S001046552300276X
-
[27]
A. Tran, J. Tranchida, T. Wildey, A. P. Thompson, Multi-fidelity machine-learning with uncertainty quantification and bayesian optimization for materials design: Ap- plication to ternary random alloys, The Journal of Chemical Physics 153 (7) (2020) 074705.arXiv:https://pubs.aip.org/ aip/jcp/article-pdf/doi/10.1063/5. 0015672/15580413/074705_1_online.pdf, ...
work page doi:10.1063/5 2020
-
[28]
J. Clérouin, P. Arnault, B.-J. Gréa, S. Guis- set, M. Vandenboomgaerde, A. J. White, L. A. Collins, J. D. Kress, C. Ticknor, Static anddynamicpropertiesofmulti-ionicplasma mixtures, Phys. Rev. E 101 (2020) 033207. doi:10.1103/PhysRevE.101.033207. URLhttps://link.aps.org/doi/10. 1103/PhysRevE.101.033207
-
[29]
G. S. Ho, V. L. Lignères, E. A. Carter, Introducing PROFESS: A new program for orbital-free density functional the- ory calculations, Comput. Phys. Com- mun. 179 (2008) 839–854.doi:https: //doi.org/10.1016/j.cpc.2008.07.002. URLhttps://www.sciencedirect. com/science/article/pii/ S0010465508002476
-
[30]
X. Shao, K. Jiang, W. Mi, A. Genova, M. Pa- vanello, DFTpy: An efficient and object- oriented platform for orbital-free DFT sim- ulations, Wiley Interdisciplinary Reviews: Computational Molecular Science 11 (2020) e1482
2020
-
[31]
W. Mi, X. Shao, C. Su, Y. Zhou, S. Zhang, Q. Li, H. Wang, L. Zhang, M. Miao, Y. Wang, Y. Ma, Atlas: A real-space finite- difference implementation of orbital-free den- sity functional theory, Comput. Phys. Com- mun. 200 (2016) 87
2016
-
[32]
Q. Xu, C. Ma, W. Mi, Y. Wang, Y. Ma, Recent advancements and challenges in orbital-free density functional theory, WIREs Computational Molecular Sci- ence 14 (3) (2024) e1724.arXiv:https: //wires.onlinelibrary.wiley.com/doi/ pdf/10.1002/wcms.1724,doi:https: //doi.org/10.1002/wcms.1724. URLhttps://wires.onlinelibrary. wiley.com/doi/abs/10.1002/wcms.1724
-
[33]
Lehtomäki, I
J. Lehtomäki, I. Makkonen, M. A. Caro, A. Harju, O. Lopez-Acevedo, Orbital-free density functional theory implementation with the projector augmented-wave method, The Journal of chemical physics 141 (2014) 234102
2014
-
[34]
Golub, S
P. Golub, S. Manzhos, CONUNDrum: A program for orbital-free density functional theory calculations, Comput. Phys. Com- mun. 256 (2020) 107365
2020
-
[35]
Gavini, K
V. Gavini, K. Bhattacharya, M. Or- tiz, Quasi-continuum orbital-free density- functional theory : A route to multi-million atom non-periodic DFT calculation, Journal of The Mechanics and Physics of Solids 55 (2007) 697
2007
-
[36]
Suryanarayana, D
P. Suryanarayana, D. Phanish, Augmented Lagrangian formulation of orbital-free den- sity functional theory, J. Comput. Phys. 275 (2014) 524
2014
-
[37]
S. Das, M. Iyer, V. Gavini, Real-space formu- lation of orbital-free density functional the- ory using finite-element discretization: The case for Al, Mg, and Al-Mg intermetallics, Physical Review B 92 (2015) 014104
2015
-
[38]
V. V. Karasiev, T. Sjostrom, S. Trickey, Finite-temperature orbital-free DFT molec- ular dynamics: Coupling Profess and Quantum Espresso, Comput. Phys. Com- mun. 185 (2014) 3240–3249.doi:https: //doi.org/10.1016/j.cpc.2014.08.023. URLhttps://www.sciencedirect. 30 com/science/article/pii/ S001046551400304X
-
[39]
F. Lambert, J. Clérouin, G. Zérah, Very-high-temperature molecular dy- namics, Phys. Rev. E 73 (2006) 016403. doi:10.1103/PhysRevE.73.016403. URLhttps://link.aps.org/doi/10. 1103/PhysRevE.73.016403
-
[40]
Lambert, Approche sans orbitale des plasmas denses, Ph.D
F. Lambert, Approche sans orbitale des plasmas denses, Ph.D. thesis, Université Paris XI, thèse de doctorat (2007). URLhttp://www.theses.fr/ 2007PA112097
2007
-
[41]
R. M. More, K. H. Warren, D. A. Young, G. B. Zimmerman, A new quotidian equation of state (QEOS) for hot dense matter, The Physics of Fluids 31 (10) (1988) 3059–3078.arXiv:https://pubs. aip.org/aip/pfl/article-pdf/31/10/ 3059/12484353/3059\_1\_online.pdf, doi:10.1063/1.866963. URLhttps://doi.org/10.1063/1.866963
-
[42]
Ayala, S
A. Ayala, S. Tomov, A. Haidar, J. Dongarra, heFFTe: Highly efficient FFT for exascale, in: V. V. Krzhizhanovskaya, G. Závodszky, M. H. Lees, J. J. Dongarra, P. M. A. Sloot, S. Brissos, J. Teixeira (Eds.), Computational Science – ICCS 2020, Springer International Publishing, Cham, 2020, pp. 262–275
2020
-
[43]
S. Cayrols, J. Li, G. Bosilca, S. Tomov, A. Ayala, J. Dongarra, Lossy all-to-all ex- change for accelerating parallel 3-D FFTs on hybrid architectures with GPUs, in: 2022 IEEE International Conference on Cluster Computing (CLUSTER), 2022, pp. 152–160. doi:10.1109/CLUSTER51413.2022.00029
-
[44]
L. W. Wang, M. P. Teter, Kinetic-energy functional of the electron density, Phys. Rev. B 45 (1992) 13196
1992
-
[45]
Stroustrup, Programming: Principles and Practice Using C++, Addison-Wesley Pro- fessional, 2014
B. Stroustrup, Programming: Principles and Practice Using C++, Addison-Wesley Pro- fessional, 2014. URLhttps://api.semanticscholar.org/ CorpusID:60689016
2014
-
[46]
R. M. Martin, Electronic Structure: Basic Theory and Practical Methods, 2nd Edition, Cambridge University Press, 2020
2020
-
[47]
M. E. Tuckerman, Statistical mechanics : theory and molecular simulation / Mark E. Tuckerman (Department of Chemistry and Courant Institute of Mathematical Sciences, New York University)., second edition. Edi- tion, Oxford graduate texts, Oxford Univer- sity Press, Oxford, United Kingdom, 2023 - 2023
2023
-
[48]
P. Minary, G. J. Martyna, M. E. Tuck- erman, Algorithms and novel applica- tions based on the isokinetic ensemble. ii. ab initio molecular dynamics, The Journal of Chemical Physics 118 (6) (2003) 2527–2538.arXiv:https://pubs. aip.org/aip/jcp/article-pdf/118/6/ 2527/19133356/2527\_1\_online.pdf, doi:10.1063/1.1534583. URLhttps://doi.org/10.1063/1. 1534583
-
[49]
E. Paquet, H. L. Viktor, Computa- tional methods for ab initio molecu- lar dynamics, Advances in Chemistry 2018 (1) (2018) 9839641.arXiv:https: //onlinelibrary.wiley.com/doi/pdf/ 10.1155/2018/9839641,doi:https: //doi.org/10.1155/2018/9839641. URLhttps://onlinelibrary.wiley.com/ doi/abs/10.1155/2018/9839641
-
[50]
R. G. Parr, W. Yang, Density-functional the- ory of atoms and molecules, Oxford Univer- sity Press, 1989
1989
-
[51]
N. D. Mermin, Thermal properties of the inhomogeneous electron gas, Phys. Rev. 137 (1965) A1441–A1443. doi:10.1103/PhysRev.137.A1441. URLhttps://link.aps.org/doi/10. 1103/PhysRev.137.A1441 31
-
[52]
F. Perrot, Gradient correction to the sta- tistical electronic free energy at nonzero temperatures: Application to equation-of- state calculations, Phys. Rev. A 20 (1979) 586–594.doi:10.1103/PhysRevA.20.586. URLhttps://link.aps.org/doi/10. 1103/PhysRevA.20.586
-
[53]
A. F. Nikiforov, V. G. Novikov, V. Uvarov, Quantum-Statistical Models of Hot Dense Matter: Methods for Computation Opac- ity and Equation of State, Birkhäuser Basel, Basel, 2005, Ch. The general- ized Thomas-Fermi model, pp. 3–28. doi:10.1007/3-7643-7346-6_1. URLhttps://doi.org/10.1007/ 3-7643-7346-6_1
-
[54]
C. F. von Weizsäcker, Zur theorie der kern- massen, Z. Phys. 96 (1935) 431
1935
-
[55]
V. V. Karasiev, T. Sjostrom, J. Dufty, S. B. Trickey, Accurate homogeneous electron gas exchange-correlation free en- ergy for local spin-density calculations, Phys. Rev. Lett. 112 (2014) 076403. doi:10.1103/PhysRevLett.112.076403. URLhttps://link.aps.org/doi/10. 1103/PhysRevLett.112.076403
-
[56]
N. Troullier, J. L. Martins, Efficient pseudopotentials for plane-wave calcula- tions, Phys. Rev. B 43 (1991) 1993–2006. doi:10.1103/PhysRevB.43.1993. URLhttps://link.aps.org/doi/10. 1103/PhysRevB.43.1993
-
[57]
M. C. Payne, M. P. Teter, D. C. Al- lan, T. A. Arias, J. D. Joannopoulos, Iterative minimization techniques for ab initio total-energy calculations: molec- ular dynamics and conjugate gradients, Rev. Mod. Phys. 64 (1992) 1045–1097. doi:10.1103/RevModPhys.64.1045. URLhttps://link.aps.org/doi/10. 1103/RevModPhys.64.1045
-
[58]
S. Morante, G. C. Rossi, M. Testa, The stress tensor of a molecular system: An exercise in statistical mechanics, The Jour- nal of Chemical Physics 125 (3) (2006) 034101.arXiv:https://pubs.aip.org/ aip/jcp/article-pdf/doi/10.1063/1. 2214719/14792114/034101_1_online.pdf, doi:10.1063/1.2214719. URLhttps://doi.org/10.1063/1. 2214719
work page doi:10.1063/1 2006
-
[59]
O. H. Nielsen, R. M. Martin, Quantum- mechanical theory of stress and force, Phys. Rev. B 32 (1985) 3780–3791. doi:10.1103/PhysRevB.32.3780. URLhttps://link.aps.org/doi/10. 1103/PhysRevB.32.3780
-
[60]
Min, A new look at the atomic level virial stress: on continuum-molecular sys- tem equivalence, Proc
Z. Min, A new look at the atomic level virial stress: on continuum-molecular sys- tem equivalence, Proc. R. Soc. Lond. A. 459 (2003) 2347–2392.doi:http://doi.org/ 10.1098/rspa.2003.1127
-
[61]
L. J. Bartolotti, R. G. Parr, The concept of pressure in density functional theory, The Journal of Chemical Physics 72 (3) (1980) 1593–1596.arXiv:https://pubs. aip.org/aip/jcp/article-pdf/72/3/ 1593/18921809/1593\_1\_online.pdf, doi:10.1063/1.439358. URLhttps://doi.org/10.1063/1.439358
-
[62]
C. E. Starrett, Thomas-fermi simulations of dense plasmas without pseudopoten- tials, Phys. Rev. E 96 (2017) 013206. doi:10.1103/PhysRevE.96.013206. URLhttps://link.aps.org/doi/10. 1103/PhysRevE.96.013206
-
[63]
T. Sjostrom, J. Daligault, Fast and ac- curate quantum molecular dynamics of dense plasmas across temperature regimes, Phys. Rev. Lett. 113 (2014) 155006. doi:10.1103/PhysRevLett.113.155006. URLhttps://link.aps.org/doi/10. 1103/PhysRevLett.113.155006
- [64]
-
[65]
Lehtola, C
S. Lehtola, C. Steigemann, M. J. Oliveira, M. A. Marques, Recent developments in libxc — a comprehensive library of function- als for density functional theory, SoftwareX 7 (2018) 1–5.doi:https://doi.org/10. 1016/j.softx.2017.11.002. URLhttps://www.sciencedirect. com/science/article/pii/ S2352711017300602
2018
-
[66]
M. J. Verstraete, J. Abreu, G. E. Alle- mand, B. Amadon, G. Antonius, M. Azizi, L. Baguet, C. Barat, L. Bastogne, R. Béjaud, J.-M.Beuken, J.Bieder, A.Blanchet, F.Bot- tin, J. Bouchet, J. Bouquiaux, E. Bousquet, J. Boust, F. Brieuc, V. Brousseau-Couture, N. Brouwer, F. Bruneval, A. Castellano, E. Castiel, J.-B. Charraud, J. Clérouin, M. Côté, C. Duval, A. ...
-
[67]
Jaupart, Statistical approach to as- trophysical flows in star forming clouds: towards the stellar initial mass function, Theses, Université de Lyon (Jul
E. Jaupart, Statistical approach to as- trophysical flows in star forming clouds: towards the stellar initial mass function, Theses, Université de Lyon (Jul. 2021). URLhttps://theses.hal.science/ tel-03367068
2021
-
[68]
A. Blanchet, F. Soubiran, M. Torrent, J. Clérouin, Extended first-principles molecular dynamics simulations of hot dense boron: equation of state and ion- ization, Contributions to Plasma Physics 62 (10) (2022) e202100234.arXiv:https: //onlinelibrary.wiley.com/doi/pdf/ 10.1002/ctpp.202100234,doi:https: //doi.org/10.1002/ctpp.202100234. URLhttps://onlineli...
-
[69]
A. Blanchet, F. Soubiran, M. Torrent, J. Clérouin, First-principles molecular- dynamics equation of state of liquid to dense plasma iron, Phys. Rev. E 111 (2025) 015206. doi:10.1103/PhysRevE.111.015206. URLhttps://link.aps.org/doi/10. 1103/PhysRevE.111.015206
-
[70]
URLhttps://www-hpc.cea.fr/en/TGCC
TGCC, supercomputing center (2026). URLhttps://www-hpc.cea.fr/en/TGCC. html 33
2026
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.