Hartree-Fock Limit for Energies in Solids
Pith reviewed 2026-06-26 16:28 UTC · model grok-4.3
The pith
Aligning radial basis functions and core orbitals with the Hartree-Fock Hamiltonian lets LAPW calculations reach the HF energy limit for solids and molecules to within a few microhartree.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
The central claim is that constructing radial basis functions and core orbitals consistently with the HF Hamiltonian removes the previous limitations of the standard LAPW treatment of nonlocal exchange, thereby allowing total energies of molecules and solids to be obtained at the Hartree-Fock limit with a precision of a few μHa.
What carries the argument
HF-consistent construction of radial basis functions and core orbitals inside the LAPW framework, which ensures proper handling of nonlocal exchange.
If this is right
- Total energies of molecules and solids are obtained to within a few μHa of the HF limit.
- Reference HF data are supplied for fourteen semiconductors and insulators.
- The standard LAPW construction remains accurate for relative energies such as molecular and solid-state formation energies and Si self-interstitial defect formation energies.
- Error control improves in hybrid-functional LAPW calculations.
- X-ray spectroscopy simulations become possible inside LAPW using hybrid-functional core orbitals.
Where Pith is reading between the lines
- The reference data can serve as benchmarks for testing basis-set convergence and pseudopotential accuracy in other all-electron or plane-wave codes.
- The same HF-consistent construction could be tested on systems with stronger correlation or larger unit cells to check whether the microhartree precision persists.
- Improved core orbitals open the possibility of direct hybrid-functional treatments of core-level spectroscopies without additional approximations.
Load-bearing premise
That building radial basis functions and core orbitals from the HF Hamiltonian itself is enough to eliminate the current limitations of standard LAPW for nonlocal exchange.
What would settle it
A comparison of the method's total energies against independently converged, exact Hartree-Fock results for the same molecules or solids that shows deviations larger than a few μHa would falsify the claimed precision.
Figures

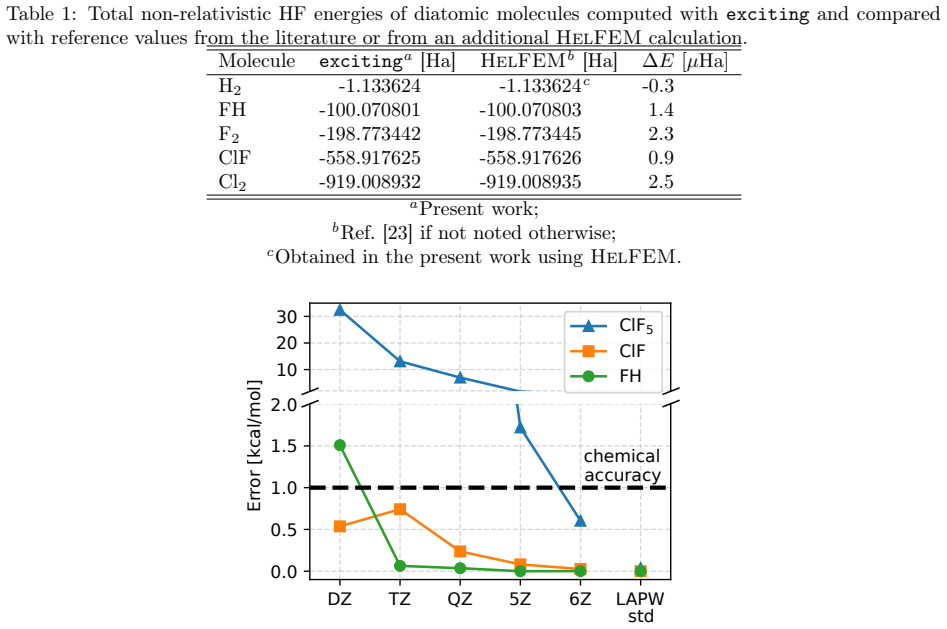
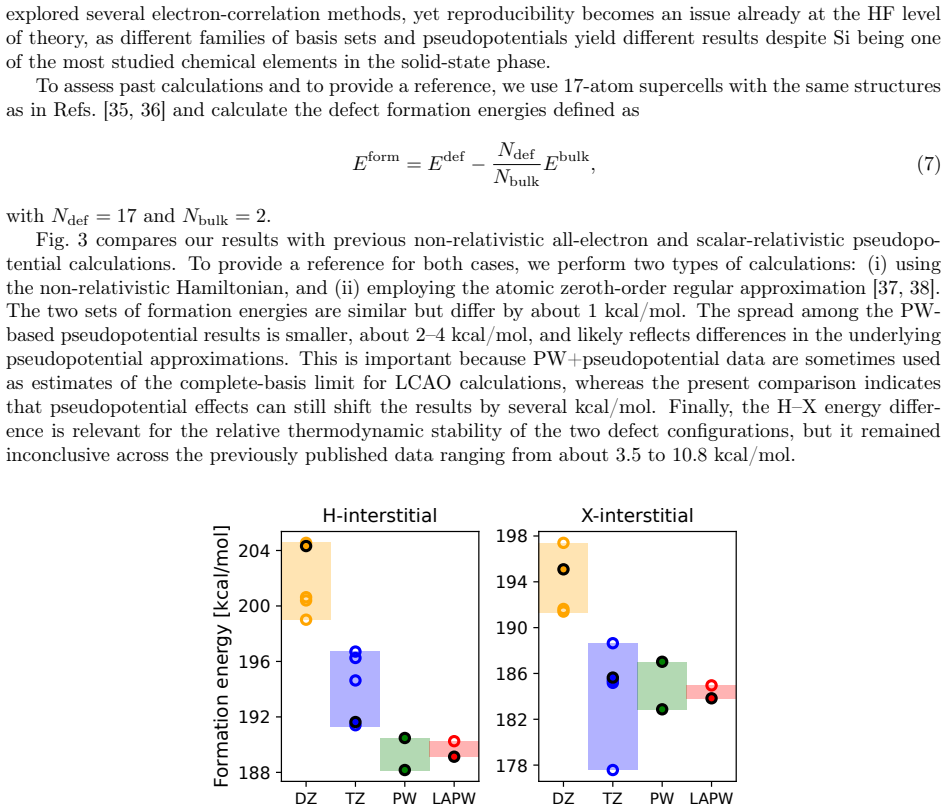
read the original abstract
This study establishes a route to the Hartree--Fock (HF) limit for molecules and solids within the linearized augmented plane wave (LAPW) framework. We remove current limitations of the standard LAPW approach to nonlocal exchange by constructing radial basis functions and core orbitals consistently with the HF Hamiltonian. The presented method yields total energies of molecules and solids with a precision of a few $\mu$Ha, and we use it to provide reference data for 14 semiconductors and insulators. For the systems considered in this study, the standard approach based on (semi)local potentials for constructing radial basis functions and core orbitals remains highly precise for practical relative energies, including molecular and solid-state formation energies and Si self-interstitial defect formation energies. More broadly, the results provide stringent all-electron benchmarks for basis-set and pseudopotential assessment, improve error control in hybrid-functional calculations within LAPW, and open the way to X-ray spectroscopy simulations within LAPW based directly on hybrid-functional core orbitals.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript presents a modification to the LAPW method in which radial basis functions and core orbitals are constructed from the HF Hamiltonian rather than from (semi)local potentials. This is claimed to remove limitations on nonlocal exchange, yielding total energies for molecules and solids at few-μHa absolute accuracy and supplying reference HF data for 14 semiconductors and insulators. The work also states that the conventional (semi)local construction remains adequate for relative energies such as formation energies and defect energies.
Significance. If the stated absolute accuracy is demonstrated, the approach would supply much-needed all-electron HF benchmarks for solids, enabling rigorous assessment of basis sets and pseudopotentials, tighter error control in hybrid-functional LAPW calculations, and direct use of hybrid core orbitals for X-ray spectroscopy simulations.
major comments (1)
- [Abstract] Abstract: the central claim of few-μHa precision for total energies and the provision of reference data for 14 materials is not accompanied by any numerical tables, convergence plots, or direct comparisons to established HF limits, leaving the accuracy assertion unverified from the manuscript text.
Simulated Author's Rebuttal
We thank the referee for their careful review and for highlighting the need for clear verification of the accuracy claims. We respond to the major comment below.
read point-by-point responses
-
Referee: [Abstract] Abstract: the central claim of few-μHa precision for total energies and the provision of reference data for 14 materials is not accompanied by any numerical tables, convergence plots, or direct comparisons to established HF limits, leaving the accuracy assertion unverified from the manuscript text.
Authors: The full manuscript contains the requested verification. Section III reports basis-set convergence studies with plots (Figs. 2–4) demonstrating that total energies converge to within a few μHa. Table I provides direct comparisons of molecular HF energies against established limits from quantum-chemistry codes. Table II lists all-electron HF total energies for the 14 semiconductors and insulators, together with comparisons to literature values where available. These data substantiate the abstract claims. Abstracts are concise summaries and conventionally omit tables and figures; the supporting evidence appears in the main text as described. revision: no
Circularity Check
No significant circularity identified
full rationale
The paper describes a direct methodological modification to the LAPW framework: radial basis functions and core orbitals are constructed from the HF Hamiltonian itself to reach the Hartree-Fock limit. This construction is presented as an implementation choice that addresses a known limitation of standard LAPW for nonlocal exchange, without any fitted parameters renamed as predictions, self-citation chains that bear the central claim, or reductions of results to inputs by definition. The reported few-μHa precision and reference data for 14 solids follow from the method's design and external benchmarks rather than internal equivalence. The derivation chain remains self-contained.
Axiom & Free-Parameter Ledger
axioms (1)
- domain assumption Standard LAPW representation of wave functions in solids remains valid when the radial basis is generated from the HF Hamiltonian.
Reference graph
Works this paper leans on
-
[1]
The Wave Mechanics of an Atom with a Non-Coulomb Central Field. Part I. Theory and Methods , volume=. Mathematical Proceedings of the Cambridge Philosophical Society , author=. 1928 , pages=. doi:10.1017/S0305004100011919 , number=
-
[2]
Fock, V. , year =. Näherungsmethode zur Lösung des quantenmechanischen Mehrkörperproblems , volume =. Zeitschrift für Physik , publisher =. doi:10.1007/bf01340294 , number =
-
[3]
Note on Hartree's Method , author =. Phys. Rev. , volume =. 1930 , month =. doi:10.1103/PhysRev.35.210.2 , url =
-
[4]
and Rothlisberger, Ursula , title =
Villard, Justin and Bircher, Martin P. and Rothlisberger, Ursula , title =. Journal of Chemical Theory and Computation , volume =. 2023 , doi =
2023
-
[5]
, title =
Hurtado, Adrian and Sekino, Hideo and Harrison, Robert J. , title =. Journal of Chemical Theory and Computation , volume =. 2024 , doi =
2024
-
[6]
Inhomogeneous Electron Gas , volume =. Phys. Rev. , author =. 1964 , pages =. doi:10.1103/physrev.136.b864 , number =
-
[7]
Self-Consistent Equations Including Exchange and Correlation Effects , volume =. Phys. Rev. , author =. 1965 , pages =. doi:10.1103/physrev.140.a1133 , number =
-
[8]
Perdew, John P. and Burke, Kieron and Ernzerhof, Matthias , year =. Generalized Gradient Approximation Made Simple , volume =. Physical Review Letters , publisher =. doi:10.1103/physrevlett.77.3865 , number =
-
[9]
2000 , doi =
An alternative way of linearizing the augmented plane-wave method , journal =. 2000 , doi =
2000
-
[10]
Linear methods in band theory , author =. Phys. Rev. B , volume =. 1975 , doi =
1975
-
[11]
Wave Functions in a Periodic Potential , author =. Phys. Rev. , volume =. 1937 , doi =
1937
-
[12]
Range-separated hybrid functionals for accurate prediction of band gaps of extended systems , volume =
Yang, Jing and Falletta, Stefano and Pasquarello, Alfredo , year =. Range-separated hybrid functionals for accurate prediction of band gaps of extended systems , volume =. npj Computational Materials , doi =
-
[13]
Physical Review B , publisher=
Gulans, Andris and Kozhevnikov, Anton and Draxl, Claudia , year=. Physical Review B , publisher=. doi:10.1103/physrevb.97.161105 , number=
-
[14]
Journal of Physics: Condensed Matter , author=
Gulans, Andris and Kontur, Stefan and Meisenbichler, Christian and Nabok, Dmitrii and Pavone, Pasquale and Rigamonti, Santiago and Sagmeister, Stephan and Werner, Ute and Draxl, Claudia , year =. exciting: a full-potential all-electron package implementing density-functional theory and many-body perturbation theory , volume =. Journal of Physics: Condense...
-
[15]
Užulis, Jānis and Sorokin, Aleksandr V. and Gulans, Andris , year =. Range-separated hybrid functionals in full-potential LAPW using adaptively compressed exchange , volume =. npj Computational Materials , publisher =. doi:10.1038/s41524-025-01733-z , number =
-
[16]
Adaptively compressed exchange in the linearized augmented plane wave formalism , author =. Phys. Rev. B , volume =. 2022 , doi =
2022
-
[17]
Journal of Physics Communications , publisher =
Užulis, Jānis and Gulans, Andris , year =. Journal of Physics Communications , publisher =. doi:10.1088/2399-6528/ac82a5 , number =
-
[18]
Simple approach to the band-structure problem , journal =. 1973 , issn =. doi:https://doi.org/10.1016/0038-1098(73)90210-X , url =
-
[19]
Numerical quality control for DFT-based materials databases , volume =
Carbogno, Christian and Thygesen, Kristian Sommer and Bieniek, Bj\". Numerical quality control for DFT-based materials databases , volume =. npj Computational Materials , publisher =. 2022 , month = apr, pages =. doi:10.1038/s41524-022-00744-4 , number =
-
[20]
Fully numerical Hartree‐Fock and density functional calculations
Lehtola, Susi , year =. Fully numerical Hartree‐Fock and density functional calculations. II. Diatomic molecules , volume =. International Journal of Quantum Chemistry , publisher =. doi:10.1002/qua.25944 , number =
-
[21]
Johnson, RD , language =. Computational Chemistry Comparison and Benchmark Database, NIST Standard Reference Database 101 , publisher =. 2002 , copyright =. doi:10.18434/T47C7Z , url =
-
[22]
and Wolfsegger, Sandra and Tew, David P
Bischoff, Florian A. and Wolfsegger, Sandra and Tew, David P. and Klopper, Wim , year =. Assessment of basis sets for F12 explicitly-correlated molecular electronic-structure methods , volume =. Molecular Physics , publisher =. doi:10.1080/00268970802708942 , number =
-
[23]
Valiev, M. and Bylaska, E.J. and Govind, N. and Kowalski, K. and Straatsma, T.P. and Van Dam, H.J.J. and Wang, D. and Nieplocha, J. and Apra, E. and Windus, T.L. and de Jong, W.A. , year =. NWChem: A comprehensive and scalable open-source solution for large scale molecular simulations , volume =. Computer Physics Communications , publisher =. doi:10.1016/...
-
[24]
Implementation of screened hybrid functionals based on the. Phys. Rev. B , author =. doi:10.1103/physrevb.83.235118 , pages =
-
[25]
Efficient calculation of the exact exchange energy in periodic systems using a truncated Coulomb potential , author =. Phys. Rev. B , volume =. 2008 , month =
2008
-
[26]
Journal of Chemical Theory and Computation , volume =
Brakestad, Anders and Jensen, Stig Rune and Tantardini, Christian and Pitteloud, Quentin and Wind, Peter and Užulis, Jānis and Gulans, Andris and Hopmann, Kathrin Helen and Frediani, Luca , title =. Journal of Chemical Theory and Computation , volume =. 2024 , doi =
2024
-
[27]
Accurate Hartree-Fock energy of extended systems using large Gaussian basis sets , author =. Phys. Rev. B , volume =. 2009 , month =. doi:10.1103/PhysRevB.80.174114 , url =
-
[28]
The Journal of Chemical Physics , volume =
Sun, Qiming , title =. The Journal of Chemical Physics , volume =. 2023 , month =. doi:10.1063/5.0155815 , url =
-
[29]
The Journal of Chemical Physics , volume =
Usvyat, Denis and Civalleri, Bartolomeo and Maschio, Lorenzo and Dovesi, Roberto and Pisani, Cesare and Schütz, Martin , title =. The Journal of Chemical Physics , volume =. 2011 , month =. doi:10.1063/1.3595514 , url =
-
[30]
Journal of Chemical Theory and Computation , volume =
Daga, Loredana Edith and Civalleri, Bartolomeo and Maschio, Lorenzo , title =. Journal of Chemical Theory and Computation , volume =. 2020 , doi =
2020
-
[31]
Gillan, M. J. and Alfè, D. and de Gironcoli, S. and Manby, F. R. , title =. Journal of Computational Chemistry , volume =. doi:https://doi.org/10.1002/jcc.21033 , url =. https://onlinelibrary.wiley.com/doi/pdf/10.1002/jcc.21033 , year =
-
[32]
Comment on ``Accurate Hartree-Fock energy of extended systems using large Gaussian basis sets'' , author =. Phys. Rev. B , volume =. 2010 , month =. doi:10.1103/PhysRevB.81.106101 , url =
-
[33]
The Journal of Chemical Physics , volume =
Stoll, Hermann and Doll, Klaus , title =. The Journal of Chemical Physics , volume =. 2012 , month =. doi:10.1063/1.3687003 , url =
-
[34]
The Journal of Chemical Physics , volume =
Grüneis, Andreas and Marsman, Martijn and Kresse, Georg , title =. The Journal of Chemical Physics , volume =. 2010 , month =. doi:10.1063/1.3466765 , url =
-
[35]
Grüneis, Andreas , year =. A coupled cluster and Møller-Plesset perturbation theory study of the pressure induced phase transition in the LiH crystal , volume =. The Journal of Chemical Physics , publisher =. doi:10.1063/1.4928645 , number =
-
[36]
Marsman, M. and Grüneis, A. and Paier, J. and Kresse, G. , year =. Second-order Møller–Plesset perturbation theory applied to extended systems. I. Within the projector-augmented-wave formalism using a plane wave basis set , volume =. The Journal of Chemical Physics , publisher =. doi:10.1063/1.3126249 , number =
-
[37]
Murnaghan, F. D. , year =. The Compressibility of Media under Extreme Pressures , volume =. Proceedings of the National Academy of Sciences , publisher =. doi:10.1073/pnas.30.9.244 , number =
-
[38]
Finite Elastic Strain of Cubic Crystals , author =. Phys. Rev. , volume =. 1947 , month =. doi:10.1103/PhysRev.71.809 , url =
-
[39]
, title =
Ye, Hong-Zhou and Berkelbach, Timothy C. , title =. Journal of Chemical Theory and Computation , volume =. 2022 , doi =
2022
-
[40]
, title =
Ye, Hong-Zhou and Berkelbach, Timothy C. , title =. Journal of Chemical Theory and Computation , volume =. 2024 , doi =
2024
-
[41]
, title =
McClain, James and Sun, Qiming and Chan, Garnet Kin-Lic and Berkelbach, Timothy C. , title =. Journal of Chemical Theory and Computation , volume =. 2017 , doi =
2017
-
[42]
and Ernzerhof, Matthias and Burke, Kieron , year =
Perdew, John P. and Ernzerhof, Matthias and Burke, Kieron , year =. Rationale for mixing exact exchange with density functional approximations , volume =. The Journal of Chemical Physics , publisher =. doi:10.1063/1.472933 , number =
-
[43]
Exploring the accuracy of the equation-of-motion coupled-cluster band gap of solids , author =. Phys. Rev. B , volume =. 2025 , month =. doi:10.1103/PhysRevB.111.L121202 , url =
-
[44]
Liao, Ke and Grüneis, Andreas , title =. The Journal of Chemical Physics , volume =. 2016 , month =. doi:10.1063/1.4964307 , url =
-
[45]
Fragment-Based Direct-Local-Ring-Coupled-Cluster Doubles Treatment Embedded in the Periodic Hartree–Fock Solution , journal =
Masur, Oliver and Sch. Fragment-Based Direct-Local-Ring-Coupled-Cluster Doubles Treatment Embedded in the Periodic Hartree–Fock Solution , journal =. 2016 , doi =
2016
-
[46]
Formation energies of silicon self-interstitials using periodic coupled cluster theory , author =. Phys. Rev. B , volume =. 2023 , month =. doi:10.1103/PhysRevB.108.115125 , url =
-
[47]
On the accuracy of numerical Hartree-Fock energies , volume =
Jensen, Frank , year =. On the accuracy of numerical Hartree-Fock energies , volume =. Theoretical Chemistry Accounts , publisher =. doi:10.1007/s00214-004-0618-8 , number =
-
[48]
The Journal of Physical Chemistry A , volume =
Maschio, Lorenzo and Kirtman, Bernard , title =. The Journal of Physical Chemistry A , volume =. 2024 , doi =
2024
-
[49]
Hybrid functionals within the all-electron
Betzinger, Markus and Friedrich, Christoph and Bl\"ugel, Stefan , journal =. Hybrid functionals within the all-electron. 2010 , publisher =
2010
-
[50]
Elimination of the linearization error and improved basis-set convergence within the FLAPW method , journal =. 2013 , issn =. doi:https://doi.org/10.1016/j.cpc.2013.07.002 , url =
-
[51]
The Elephant in the Room of Density Functional Theory Calculations , journal =
Jensen, Stig Rune and Saha, Santanu and Flores-Livas, Jos. The Elephant in the Room of Density Functional Theory Calculations , journal =. 2017 , doi =
2017
-
[52]
Advanced Theory and Simulations , volume =
Vona, Cecilia and Nabok, Dmitrii and Draxl, Claudia , title =. Advanced Theory and Simulations , volume =. doi:https://doi.org/10.1002/adts.202100496 , url =. https://advanced.onlinelibrary.wiley.com/doi/pdf/10.1002/adts.202100496 , year =
-
[53]
Singh and Lars Nordstr
David J. Singh and Lars Nordstr. Planewaves, Pseudopotentials, and the. 2006 , isbn =
2006
-
[54]
2018 , howpublished =
Susi Lehtola , title =. 2018 , howpublished =
2018
-
[55]
and Casassa, Silvia and Civalleri, Bartolomeo and Donà, Lorenzo and Bush, Ian J
Erba, Alessandro and Desmarais, Jacques K. and Casassa, Silvia and Civalleri, Bartolomeo and Donà, Lorenzo and Bush, Ian J. and Searle, Barry and Maschio, Lorenzo and Edith-Daga, Loredana and Cossard, Alessandro and Ribaldone, Chiara and Ascrizzi, Eleonora and Marana, Naiara L. and Flament, Jean-Pierre and Kirtman, Bernard , title =. Journal of Chemical T...
2023
-
[56]
and Iannuzzi, Marcella and Del Ben, Mauro and Rybkin, Vladimir V
Kühne, Thomas D. and Iannuzzi, Marcella and Del Ben, Mauro and Rybkin, Vladimir V. and Seewald, Patrick and Stein, Frederick and Laino, Teodoro and Khaliullin, Rustam Z. and Schütt, Ole and Schiffmann, Florian and Golze, Dorothea and Wilhelm, Jan and Chulkov, Sergey and Bani-Hashemian, Mohammad Hossein and Weber, Valéry and Borštnik, Urban and Taillefumie...
-
[57]
M. J. Frisch and G. W. Trucks and H. B. Schlegel and G. E. Scuseria and M. A. Robb and J. R. Cheeseman and G. Scalmani and V. Barone and G. A. Petersson and H. Nakatsuji and X. Li and M. Caricato and A. V. Marenich and J. Bloino and B. G. Janesko and R. Gomperts and B. Mennucci and H. P. Hratchian and J. V. Ortiz and A. F. Izmaylov and J. L. Sonnenberg an...
2016
-
[58]
and Teukolsky, Saul A
Press, William H. and Teukolsky, Saul A. and Vetterling, William T. and Flannery, Brian P. , title =. 2007 , isbn =
2007
-
[59]
Dovesi, Roberto and Doll, Klaus and Causà, Mauro and R. The Hartree–Fock Exchange for Crystalline Systems: The Implementation with an (All-Electron) Gaussian-Type Basis Set and Numerical Evidence with Reference to Perovskites , journal =. 2026 , doi =
2026
-
[60]
Transcorrelated wave-function framework for solids: An application to bulk and defected silicon , author =. Phys. Rev. B , volume =. 2026 , month =. doi:10.1103/d65l-5865 , url =
-
[61]
The Journal of Chemical Physics , volume=
Relativistic regular two-component Hamiltonians , author=. The Journal of Chemical Physics , volume=. 1993 , publisher=
1993
-
[62]
The Journal of Chemical Physics , volume=
Relativistic total energy using regular approximations , author=. The Journal of Chemical Physics , volume=. 1994 , publisher=
1994
-
[63]
Robert J. Harrison and George I. Fann and Takeshi Yanai and Zhengting Gan and Gregory Beylkin , title =. 2004 , month = dec, publisher =. doi:10.1063/1.1791051 , url =
-
[64]
Lejaeghere, K. And Bihlmayer, G. And Bjorkman, T. And Blaha, P. And Blugel, S. And Blum, V. And Caliste, D. And Castelli, I. E. And Clark, S. J. And Dal Corso, A. And et al. , year=. Science , publisher=. doi:10.1126/science.aad3000 , number=
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.