Consistent Initial States with Constant Circuit Depth for Quantum Computational Chemistry
Pith reviewed 2026-06-26 00:44 UTC · model grok-4.3
The pith
Separable pair approximation states deliver consistent quantum chemistry results at constant circuit depth.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
Separable pair approximations supply consistent initial states for variational quantum eigensolvers. When prepared in an orbital-optimized framework they yield approximations of hydrogen chains, alkanes, and small molecules whose accuracy and classical cost track those of Hartree-Fock, all while requiring only constant-depth circuits with linear gate count and parameter count.
What carries the argument
Separable pair approximation (SPA) states, which compile to shallow constant-depth circuits, remain classically simulable, and integrate into larger variational procedures.
If this is right
- SPA states can serve as standalone methods or as subroutines inside larger quantum algorithms without raising circuit depth.
- Classical simulability removes most variational quantum algorithm bottlenecks for these initial states.
- The states supply black-box applicability with performance comparable to Hartree-Fock across the tested molecular classes.
- SPA circuits remain chemically motivated low-depth building blocks that combine with existing subspace and circuit strategies.
Where Pith is reading between the lines
- If the pattern holds for larger systems, SPA states could replace deeper ansatze in near-term devices where noise limits circuit depth.
- Their linear parameter count may allow systematic improvement by adding more pairs without exponential growth in classical optimization cost.
- Because the states are exactly solvable classically, any discrepancy between SPA and full configuration interaction can be attributed directly to the pair approximation rather than optimization failure.
Load-bearing premise
The chosen test systems and orbital-optimized VQE procedure are representative enough to establish general consistency of SPA states.
What would settle it
A single additional molecule outside the tested families where SPA energies deviate markedly from Hartree-Fock while keeping the same circuit depth would falsify the consistency claim.
Figures

read the original abstract
Variational quantum eigensolvers have been extensively studied, yet there are still no methods that offer black-box applicability with consistent performance. Separable pair approximations promise to be candidates for such methods: they compile to shallow constant-depth quantum circuits with linear gate count and parameter dependence and circumvent most bottlenecks of variational quantum algorithms through their classical simulability. At the same time, they seamlessly integrate into prominent more general circuit designs and subspace strategies. So far, their capability as a consistent method has only been indicated and demonstrations have been restricted to manually designed model systems. In this work, we extensively evaluate the consistency of SPA states for hydrogen chains, alkanes, and small molecules within an orbital-optimized VQE framework. Our benchmarks demonstrate consistent approximations with classical complexity comparable to Hartree-Fock. Our open-source implementation within the Tequila framework allows convenient use of the algorithms as a standalone method or as a subpart of more extensive procedures. Our results underpin the potential of SPA circuits as scalable, chemically motivated low-depth circuits with various applications and validate their usage as a chemically consistent method.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript evaluates separable pair approximation (SPA) states as initial states for variational quantum eigensolvers in quantum computational chemistry. It reports benchmarks on hydrogen chains, alkanes, and small molecules within an orbital-optimized VQE framework, claiming that these states produce consistent approximations whose classical computational cost is comparable to Hartree-Fock. The work emphasizes the constant circuit depth, linear gate count, and classical simulability of SPA circuits, their integration potential with other methods, and provides an open-source Tequila implementation.
Significance. If the benchmark results are robust, the paper supplies concrete evidence that SPA states can serve as a scalable, chemically motivated low-depth circuit ansatz with practical classical complexity matching Hartree-Fock. The explicit grounding in classical simulability, the open-source code release, and the demonstration of seamless integration into VQE procedures are strengths that support reproducibility and broader applicability in quantum chemistry simulations.
minor comments (3)
- The abstract states that benchmarks demonstrate consistent approximations but does not include any quantitative metrics, error bars, or baseline comparisons; adding one or two representative numerical results (e.g., energy errors or timing ratios) would strengthen the summary.
- The manuscript would benefit from a brief explicit definition or operational criterion for 'consistency' of the SPA approximations (e.g., bounded error relative to a reference method across the test set) to make the central claim easier to evaluate.
- Figure captions and axis labels should be checked for completeness so that readers can interpret the benchmark plots without returning to the main text.
Simulated Author's Rebuttal
We thank the referee for their positive assessment of our work on separable pair approximation (SPA) states as initial states for orbital-optimized VQE. The referee summary correctly identifies the benchmarks on hydrogen chains, alkanes, and small molecules, the constant circuit depth, linear gate count, classical simulability, and the open-source Tequila implementation. No specific major comments were raised in the report.
Circularity Check
No circularity in derivation chain; claims rest on independent benchmarks
full rationale
The paper's core assertions concern new benchmark results for SPA states on hydrogen chains, alkanes, and small molecules inside an orbital-optimized VQE setting. These evaluations are presented as fresh computations whose outcomes are not forced by any internal definitions, fitted parameters renamed as predictions, or self-citation chains. Classical simulability of SPA states is invoked as an enabling property with an accompanying open-source Tequila implementation, allowing external verification. No load-bearing step in the reported consistency reduces by construction to prior inputs or self-referential assumptions; the work therefore qualifies as self-contained against external benchmarks.
Axiom & Free-Parameter Ledger
axioms (1)
- standard math Standard quantum mechanical variational principles and orbital optimization assumptions underlying VQE
Reference graph
Works this paper leans on
-
[1]
Fig- ure 1 compares the SPA dissociation curve against HF, CCSD(T), and FCI
Dissociation of linear H6 We first examine the dissociation behaviour of SPA usingH 6 as a representative benchmark system. Fig- ure 1 compares the SPA dissociation curve against HF, CCSD(T), and FCI. 0.5 1.0 1.5 2.0 2.5 3.0 3.5 Interatomic distance (Å) 3.2 3.0 2.8 2.6 2.4 2.2 Energy (Hartree) SPA HF CCSD(T) FCI Figure 1: Dissociation energy curves of lin...
-
[2]
Figure 2 (top) shows the energy error relative to FCI as a function of bond length
Scaling with system size (H4–H10) We next consider hydrogen chainsH4 toH 10 to study system-size scaling across the full dissociation coordi- nate. Figure 2 (top) shows the energy error relative to FCI as a function of bond length. Near equilibrium, the energy error is approximately 15, 29, 43, and 59 mHa forH4,H 6,H 8, andH10, respec- tively. At intermed...
-
[3]
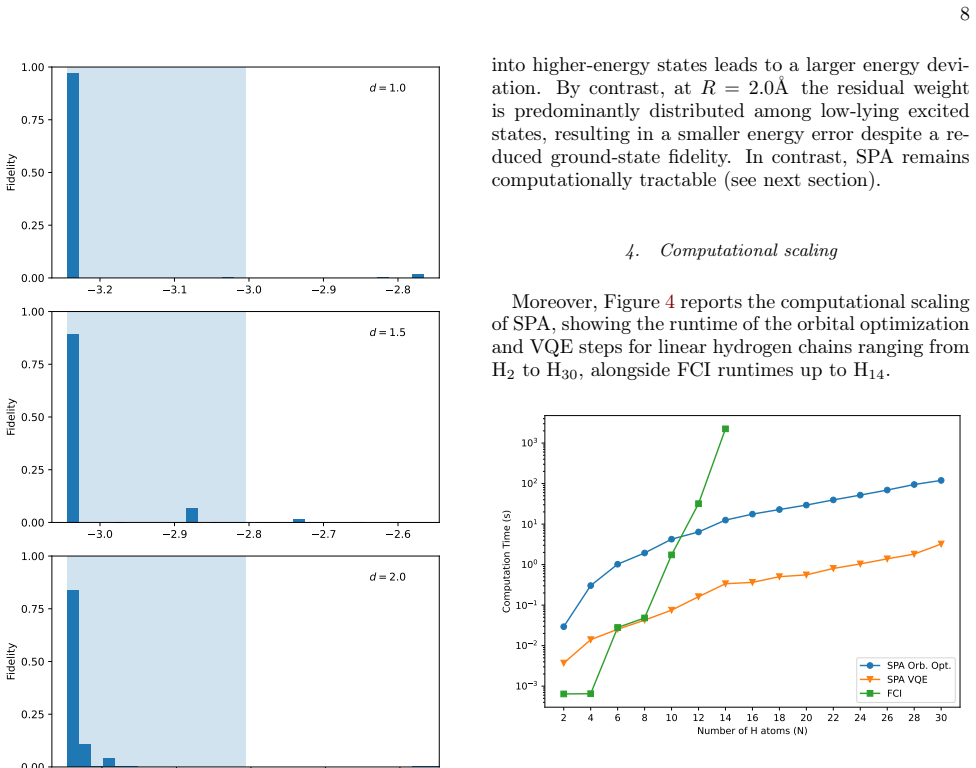
The blue shaded region indicates the UV–vis excitation window relative to the ground state
Wavefunction analysis via fidelity spectra To gain further insight into the origin of the observed energy errors, we analyze the distribution of SPA wave- 8 3.2 3.1 3.0 2.9 2.8 0.00 0.25 0.50 0.75 1.00Fidelity d = 1.0 3.0 2.9 2.8 2.7 2.6 0.00 0.25 0.50 0.75 1.00Fidelity d = 1.5 2.8 2.7 2.6 2.5 2.4 Eigenenergy (Hartree) 0.00 0.25 0.50 0.75 1.00Fidelity d =...
-
[4]
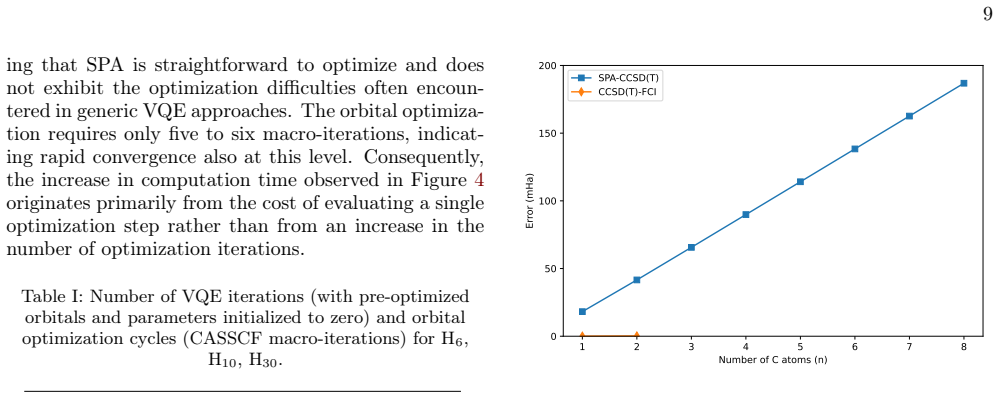
2 4 6 8 10 12 14 16 18 20 22 24 26 28 30 Number of H atoms (N) 10 3 10 2 10 1 100 101 102 103 Computation Time (s) SPA Orb
Computational scaling Moreover, Figure 4 reports the computational scaling of SPA, showing the runtime of the orbital optimization and VQE steps for linear hydrogen chains ranging from H2 toH 30, alongside FCI runtimes up toH14. 2 4 6 8 10 12 14 16 18 20 22 24 26 28 30 Number of H atoms (N) 10 3 10 2 10 1 100 101 102 103 Computation Time (s) SPA Orb. Opt....
-
[5]
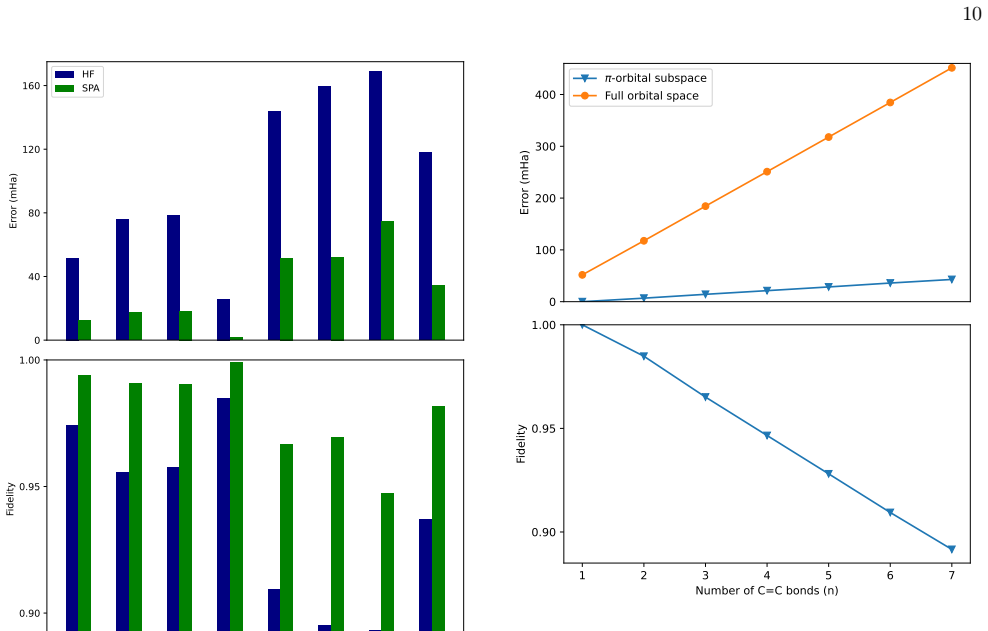
The error increases ap- proximately linearly with molecular size, analogous to that observed previously for the hydrogen chains
Alkanes Figure 5 shows the SPA energy error relative to CCSD(T) for the alkane series. The error increases ap- proximately linearly with molecular size, analogous to that observed previously for the hydrogen chains. For CH4 andC 2H6, CCSD(T)isinexcellentagreementwith FCI, with negligible energy differences, justifying its use as a reference for larger alk...
-
[6]
SPA consistently improves upon HF across all systems, although never reaching chemical accuracy (1.6 mHa) except for hydrogen fluoride
Small molecules The top panel of Figure 6 compares SPA and HF en- ergy errors relative to FCI for a set of small molecules. SPA consistently improves upon HF across all systems, although never reaching chemical accuracy (1.6 mHa) except for hydrogen fluoride. For molecules containing only single bonds, the SPA energy error remains below 35mHa, while syste...
-
[7]
Figure 7 (top) shows the SPA energy error relative to CCSD(T) in the full orbital space
Conjugatedπ-systems ToassessSPAinthepresenceofdelocalizedelectrons, we consider the conjugated polyene seriesC2nH2n+2 up ton= 7. Figure 7 (top) shows the SPA energy error relative to CCSD(T) in the full orbital space. To iso- late the role of the delocalizedπelectrons, we further performed calculations in active spaces containing only theπorbitals, for wh...
-
[8]
Y. Cao, J. Romero, J. P. Olson, M. Degroote, P. D. Johnson, M. Kieferová, I. D. Kivlichan, T. Menke, B. Peropadre, N. P. Sawaya,et al., Chem. Rev.119, 10856 (2019)
2019
-
[9]
Yuan, Rev
S.McArdle, S.Endo, A.Aspuru-Guzik, S.C.Benjamin, and X. Yuan, Rev. Mod. Phys.92, 015003 (2020)
2020
-
[10]
A.Aspuru-Guzik, A.D.Dutoi, P.J.Love,andM.Head- Gordon, Science309, 1704 (2005)
2005
-
[11]
Reiher, N
M. Reiher, N. Wiebe, K. M. Svore, D. Wecker, and M. Troyer, Proceedings of the Na- tional Academy of Sciences114, 7555 (2017), https://www.pnas.org/content/114/29/7555.full.pdf
2017
-
[12]
R. Babbush, R. King, S. Boixo, W. Huggins, T. Khat- tar, G. H. Low, J. R. McClean, T. O’Brien, and N. C. Rubin, The grand challenge of quantum applications (2025), arXiv:2511.09124 [quant-ph]
arXiv 2025
-
[13]
Caesura, C
A. Caesura, C. L. Cortes, W. Pol, S. Sim, M. Steudtner, G.-L. R. Anselmetti, M. Degroote, N. Moll, R. Santa- gati, M. Streif, and C. S. Tautermann, PRX Quantum 6, 030337 (2025)
2025
-
[14]
K. Gratsea and M. Otten, Achieving utility-scale ap- plications through full stack co-design of fault tolerant quantum computers (2025), arXiv:2510.26547 [quant- ph]
arXiv 2025
-
[15]
S. Lee, J. Lee, H. Zhai,et al., Nature Communications 14, 1952 (2023)
1952
-
[16]
Louvet, T
T. Louvet, T. Ayral, and X. Waintal, Phys. Rev. B113, 125112 (2026)
2026
-
[17]
P. W. Anderson, Phys. Rev. Lett.18, 1049 (1967)
1967
-
[18]
Peruzzo, J
A. Peruzzo, J. McClean, P. Shadbolt, M.-H. Yung, X.-Q. Zhou, P. J. Love, A. Aspuru-Guzik, and J. L. O’brien, Nature Communications5, 4213 (2014)
2014
-
[19]
J. R. McClean, J. Romero, R. Babbush, and A. Aspuru- Guzik, New Journal of Physics18, 023023 (2016)
2016
-
[20]
Bharti, A
K. Bharti, A. Cervera-Lierta, T. H. Kyaw, T. Haug, S. Alperin-Lea, A. Anand, M. Degroote, H. Heimonen, J. S. Kottmann, T. Menke,et al., Reviews of Modern Physics94, 015004 (2022)
2022
-
[21]
J. F. Gonthier, M. D. Radin, C. Buda, E. J. Doskocil, C. M. Abuan, and J. Romero, Identifying challenges to- wards practical quantum advantage through resource estimation: the measurement roadblock in the vari- ational quantum eigensolver (2020), arxiv:2012.04001 [quant-ph]
arXiv 2020
-
[22]
S. Patel, P. Jayakumar, T.-C. Yen, and A. F. Izmaylov, Chemical Reviews125, 7490 (2025), pMID: 40690271, https://doi.org/10.1021/acs.chemrev.5c00055
-
[23]
J. R. McClean, S. Boixo, V. N. Smelyanskiy, R. Bab- bush, and H. Neven, Nat. Commun.9, 1 (2018)
2018
-
[24]
Romero, R
J. Romero, R. Babbush, J. R. McClean, C. Hempel, P. J. Love, and A. Aspuru-Guzik, Quantum Science and Technology4, 014008 (2019)
2019
-
[25]
Anand, P
A. Anand, P. Schleich, S. Alperin-Lea, P. W. K. Jensen, S. Sim, M. Díaz-Tinoco, J. S. Kottmann, M. Degroote, A.F.Izmaylov,andA.Aspuru-Guzik,ChemicalSociety Reviews51, 1659 (2022)
2022
-
[26]
M. Krompiec, J. J. M. Kirsopp, A. M. Romero, and V. P. Soloviev, Journal of Chemical Theory and Computation22, 257 (2026), pMID: 41452946, https://doi.org/10.1021/acs.jctc.5c01452
-
[27]
Patel, P
S. Patel, P. Jayakumar, R. Huang, T. Zeng, and A. F. Izmaylov, Journal of Chemical Theory and Computa- tion22, 3937 (2026)
2026
-
[28]
H. R. Grimsley, S. E. Economou, E. Barnes, and N. J. Mayhall, Nature Communications10, 1 (2019)
2019
-
[29]
I. G. Ryabinkin, T.-C. Yen, S. N. Genin, and A. F. Izmaylov, J. Chem. Theory Comput.14, 6317 (2018)
2018
-
[30]
R. A. Lang, I. G. Ryabinkin, and A. F. Izmaylov, arxiv:2002.05701 (2020)
arXiv 2002
-
[31]
N. H. Stair, R. Huang, and F. A. Evangelista, J. Chem. Theory Comput.16, 2236 (2020)
2020
-
[32]
J. S. Kottmann and F. Scala, Journal of Chemical The- ory and Computation20, 3514 (2024)
2024
-
[33]
J. Park, C. C. Frink, and M. Otten, arXiv preprint arXiv:2603.13188 (2026)
arXiv 2026
-
[34]
H. G. A. Burton, D. Marti-Dafcik, D. P. Tew, and D. J. Wales, Nature 10.48550/arxiv.2207.00085 (2022)
-
[35]
J. S. Kottmann, Quantum7, 1073 (2023)
2023
-
[36]
S. E. Ghasempouri, G. W. Dueck, and S. D. Baerdemacker, Modular cluster circuits for the vari- ational quantum eigensolver (2023), arXiv:2305.04425 [quant-ph]
arXiv 2023
-
[37]
H. G. A. Burton, Physical Review Research6, 023300 (2024)
2024
-
[38]
Marti-Dafcik, H
D. Marti-Dafcik, H. G. A. Burton, and D. P. Tew, Phys. Rev. Res.7, 013191 (2025)
2025
-
[39]
J. S. Kottmann and A. Aspuru-Guzik, Physical Re- view A105, 032449 (2022), arxiv:2105.03836 [physics, physics:quant-ph]
arXiv 2022
-
[40]
Weber, A
M. Weber, A. Anand, A. Cervera-Lierta, J. S. Kottmann, T. H. Kyaw, B. Li, A. Aspuru-Guzik, C. Zhang, and Z. Zhao, Physical Review Research4, 033217 (2022)
2022
-
[41]
P. Schleich, J. S. Kottmann, and A. Aspuru-Guzik, Im- proving the accuracy of the variational quantum eigen- solver for molecular systems by the explicitly-correlated perturbative [2]R12-correction (2021), arxiv:2110.06812 [quant-ph]. 12
arXiv 2021
-
[42]
P. Schleich, J. Boen, L. Cincio, A. Anand, J. S. Kottmann, S. Tretiak, P. A. Dub, and A. Aspuru-Guzik, Journal of Chemical Theory and Computation19, 4952 (2023), pMID: 37490516, https://doi.org/10.1021/acs.jctc.3c00335
-
[43]
Bincoletto, K
D. Bincoletto, K. Stein, J. Motyl, and J. S. Kottmann, Machine Learning: Science and Technology7, 035055 (2026)
2026
-
[44]
F. J. de Arco Santos and J. S. Kottman, Quantum Science and Technology 10.1088/2058-9565/adbdee (2025)
-
[45]
E. S. Gil, M. Oppel, J. S. Kottmann, and L. González, Chemical Science16, 596 (2025)
2025
-
[46]
J. S. Kottmann, P. Schleich, T. Tamayo-Mendoza, and A. Aspuru-Guzik, The Journal of Physical Chemistry Letters12, 663 (2021)
2021
-
[47]
V. E. Elfving, M. Millaruelo, J. A. Gámez, and C. Gogolin, Physical Review A103, 032605 (2021)
2021
-
[48]
L. Zhao, J. Goings, K. Wright, J. Nguyen, J. Kim, S. Johri, K. Shin, W. Kyoung, J. I. Fuks, J.-K. K. Rhee, and Y. M. Rhee, npj Quantum Information9, 60 (2023)
2023
-
[49]
T. Y. Nikolaienko and L. A. Bulavin, International Journal of Quantum Chemistry119, e25798 (2019)
2019
-
[50]
J. S. Kottmann, S. Alperin-Lea, T. Tamayo-Mendoza, A. Cervera-Lierta, C. Lavigne, T.-C. Yen, V. Vertelet- skyi, P. Schleich, A. Anand, M. Degroote, S. Chaney, M. Kesibi, N. G. Curnow, B. Solo, G. Tsilimigkounakis, C. Zendejas-Morales, A. F. Izmaylov, and A. Aspuru- Guzik, Quantum Science and Technology6, 024009 (2021)
2021
-
[51]
F. J. del Arco Santos, D. Bincoletto, J. S. Kottmann, and N. Roshani, Project-sunrise (2026), github Repos- itory
2026
-
[52]
Suzuki, Y
Y. Suzuki, Y. Kawase, Y. Masumura, Y. Hiraga, M. Nakadai, J. Chen, K. M. Nakanishi, K. Mi- tarai, R. Imai, S. Tamiya, T. Yamamoto, T. Yan, T. Kawakubo, Y. O. Nakagawa, Y. Ibe, Y. Zhang, H. Yamashita, H. Yoshimura, A. Hayashi, and K. Fujii, Quantum5, 559 (2021)
2021
-
[53]
McClean, N
J. McClean, N. Rubin, K. Sung, I. D. Kivlichan, X. Bonet-Monroig, Y. Cao, C. Dai, E. S. Fried, C. Gid- ney, B. Gimby,et al., Quantum Science and Technology (2020)
2020
-
[54]
Q. Sun, T. C. Berkelbach, N. S. Blunt, G. H. Booth, S. Guo, Z. Li, J. Liu, J. D. McClain, E. R. Sayfutyarova, S. Sharma, S. Wouters, and G. K.-L. Chan, Wiley In- terdisciplinary Review Compututational Molecular Sci- ence8, e1340 (2018)
2018
-
[55]
T. Y. Nikolaienko, L. A. Bulavin, and D. M. Hovorun, Computational and Theoretical Chemistry1050, 15 (2014)
2014
-
[56]
Barta, github:lily-barta/SPA_Benchmark (2026)
L. Barta, github:lily-barta/SPA_Benchmark (2026)
2026
-
[57]
L. Zhao, Q. Wang, J. J. Goings, K. Shin, W. Kyoung, S. Noh, Y. M. Rhee, and K. Kim, npj Quantum Infor- mation10, 76 (2024)
2024
-
[58]
P. A. Limacher, The Journal of Chemical Physics164 (2026)
2026
-
[59]
L. van der Horst, M. Periyasamy, A. Y. Dubey, D. Bincoletto, J. S. Kottmann, and D. D. Scherer, (submitted to IEEE Quantum Week 2026) (2026), arXiv:2605.04174 [quant-ph]
Pith/arXiv arXiv 2026
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.