Shallow Quantum Circuits for Deep Chemistry via Valence Bond Embeddings
Pith reviewed 2026-06-26 04:51 UTC · model grok-4.3
The pith
Valence bond embeddings with hybrid encodings produce shallow circuits for full-molecule variational quantum chemistry.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
Embedding the molecular electronic structure in a structured valence-bond basis and mapping it through a hybrid fermionic-bosonic encoding yields a shallow, hardware-executable circuit whose variational minimum recovers ground-state energies that match or exceed the accuracy of active-space restricted circuits on the same hardware budget.
What carries the argument
Structured valence bond embedding combined with hybrid Fermionic-Bosonic encodings, which directly maps the full molecular Hamiltonian into a variational ansatz circuit without active-space truncation.
If this is right
- VQE calculations become possible on molecular sizes previously excluded by active-space size limits.
- The circuits supply initial states for projective algorithms such as quantum phase estimation on the complete system.
- Dynamical observables can be computed directly from the full Hamiltonian using the same circuit family.
- Ground-state approximations remain accurate relative to exact solutions across the tested chemically relevant molecules.
Where Pith is reading between the lines
- The same embedding structure could be reused for time-dependent simulations if the valence-bond basis preserves the required time-evolution symmetries.
- Extending the method to larger basis sets might reduce reliance on separate basis-set extrapolation procedures.
- The circuit construction could be paired with existing error-mitigation techniques to improve results on current noisy devices without changing the core ansatz.
Load-bearing premise
The variational minimum reached by the circuit remains close to the exact ground-state energy when the full molecular system is encoded without active-space cuts or extra error-mitigation steps.
What would settle it
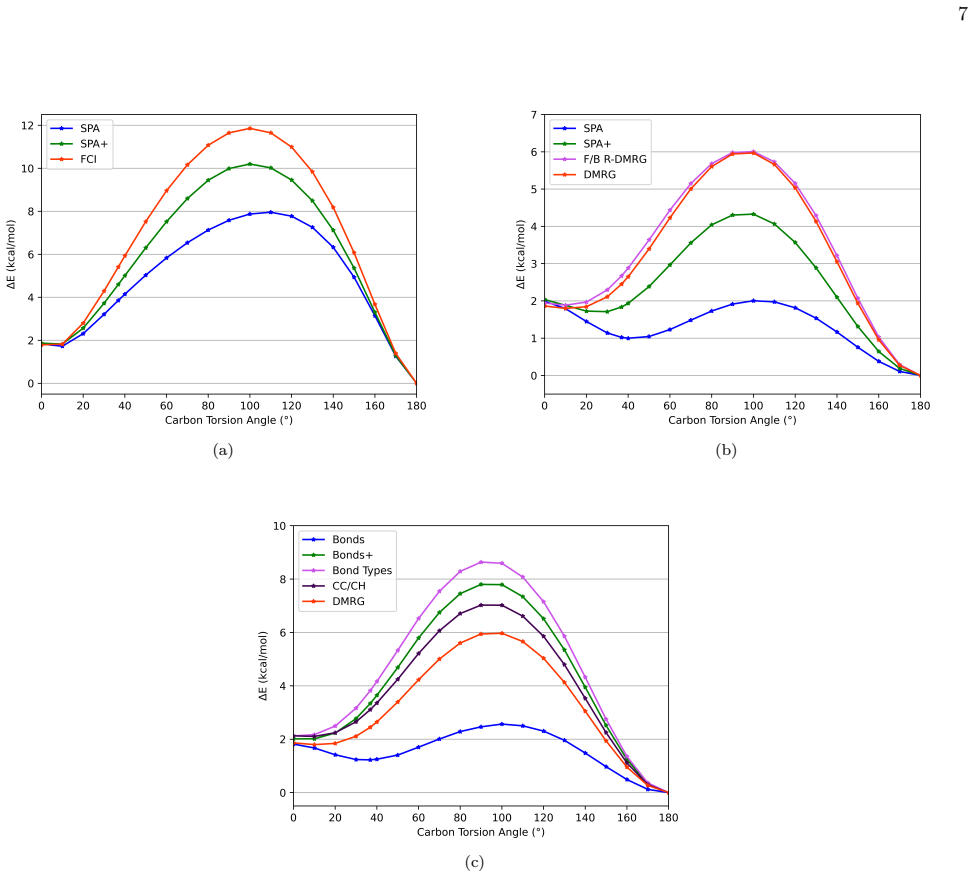
Optimizing the circuit on a molecule such as water in a double-zeta basis and finding that the final energy lies more than 1.6 millihartree above the exact full-configuration-interaction value.
Figures
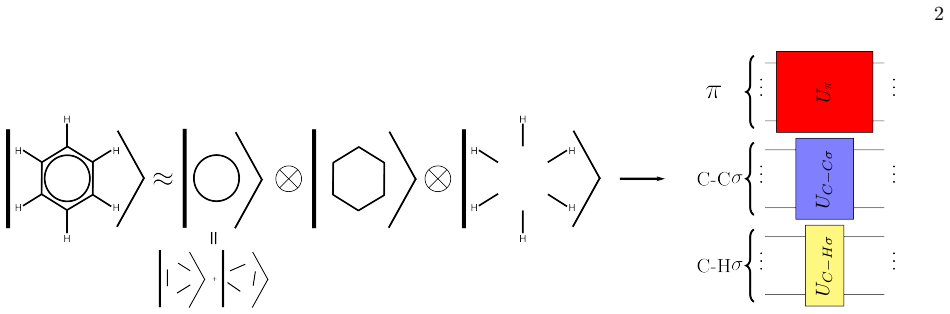
read the original abstract
Quantum chemistry is one of the major potential applications in quantum computation. Currently there is a considerable focus on relatively small active spaces as a consequence of hardware noise and exponential bottlenecks in simulations. In the long run, there will be an increasing demand in reliable approximations for larger systems -- both, as initial states for projective algorithms like the quantum phase estimation or for the evaluation of dynamical properties. While numerous approaches to select active spaces and extrapolate basis set accuracy exist, there is currently no consistent approach that results in a single quantum circuit for the total system. In this work, we combine hybrid Fermionic-Bosonic encodings with the structured approach of Quantum Valence Bond Theory to directly construct quantum circuits for comparably large molecular systems. With this approach we are able to push simulability barrier of variational quantum eigensolvers towards chemically relevant systems and demonstrate circuit designs that outperform active space counterparts and achieve good approximations with respect to the exact solutions.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript proposes combining hybrid Fermionic-Bosonic encodings with the structured valence-bond approach of Quantum Valence Bond Theory to construct shallow quantum circuits for variational quantum eigensolvers on molecular systems. It claims this enables direct treatment of the total system (without active-space restrictions), pushes the simulability barrier for VQE toward chemically relevant molecules, and yields circuits that outperform active-space counterparts while achieving good agreement with exact solutions.
Significance. If the central construction is shown to produce variational minima close to the exact ground state of the full molecular Hamiltonian, the work would provide a systematic route to single-circuit ansatze for larger systems, which could serve as useful initial states for projective methods and extend practical VQE applicability.
major comments (2)
- [Valence-bond embedding and hybrid encoding sections] The stress-test concern lands: Quantum Valence Bond Theory relies on a structured selection of valence configurations and bond embeddings. Unless the manuscript proves (e.g., via explicit span argument or amplitude bounds) that the resulting ansatz covers the full configuration space of the molecular Hamiltonian or that omitted sectors have rigorously zero weight, the variational minimum is guaranteed only for the embedded subspace. This directly undermines the claim of operating on the 'total system' without effective truncation equivalent to an active-space restriction. The relevant discussion appears in the sections describing the valence-bond embedding and the hybrid encoding construction.
- [Results and numerical validation sections] The abstract states that the circuits 'achieve good approximations with respect to the exact solutions' and 'outperform active space counterparts,' yet the provided text contains no numerical benchmarks, circuit diagrams, or derivation steps. If the full manuscript likewise lacks concrete energy errors, circuit depths, or comparisons on specific molecules (e.g., against FCI or active-space VQE), the performance claims cannot be assessed and the central assertion remains unverified.
minor comments (2)
- [Introduction] Clarify the precise definition of 'total system' versus the embedded model in the introduction and methods; the current phrasing risks conflating the two.
- [Methods] Add explicit statements on the number of free parameters retained after the valence-bond embedding; the abstract asserts a 'consistent approach' but does not address whether any implicit fitting remains.
Simulated Author's Rebuttal
We thank the referee for the careful and constructive review. We address each major comment below and commit to revisions that clarify the scope of the ansatz and substantiate the performance claims with explicit data.
read point-by-point responses
-
Referee: [Valence-bond embedding and hybrid encoding sections] The stress-test concern lands: Quantum Valence Bond Theory relies on a structured selection of valence configurations and bond embeddings. Unless the manuscript proves (e.g., via explicit span argument or amplitude bounds) that the resulting ansatz covers the full configuration space of the molecular Hamiltonian or that omitted sectors have rigorously zero weight, the variational minimum is guaranteed only for the embedded subspace. This directly undermines the claim of operating on the 'total system' without effective truncation equivalent to an active-space restriction.
Authors: We agree that the ansatz is defined within the valence-bond embedded subspace and that the manuscript provides no explicit span argument or amplitude bounds showing that omitted sectors have zero weight. The variational minimum is therefore guaranteed only within this subspace. We will revise the valence-bond embedding and hybrid encoding sections to remove any implication of unrestricted full-system treatment and instead describe the approach as a structured subspace approximation designed to capture dominant correlations, with explicit discussion of its relation to active-space restrictions. revision: yes
-
Referee: [Results and numerical validation sections] The abstract states that the circuits 'achieve good approximations with respect to the exact solutions' and 'outperform active space counterparts,' yet the provided text contains no numerical benchmarks, circuit diagrams, or derivation steps. If the full manuscript likewise lacks concrete energy errors, circuit depths, or comparisons on specific molecules (e.g., against FCI or active-space VQE), the performance claims cannot be assessed and the central assertion remains unverified.
Authors: The referee is correct: if the reviewed manuscript lacks numerical benchmarks, the abstract claims cannot be assessed. We will add a dedicated results section containing explicit energy errors relative to exact (FCI) solutions, circuit depths, and direct comparisons against active-space VQE on specific molecules to verify the stated performance. revision: yes
Circularity Check
No circularity; derivation self-contained from external encodings and valence-bond theory
full rationale
The provided abstract and text describe combining hybrid Fermionic-Bosonic encodings with Quantum Valence Bond Theory to build circuits for larger systems. No equations, fitted parameters, or self-citations are shown that reduce any claimed prediction or uniqueness result to the inputs by construction. The performance claims are presented as outcomes of the circuit construction rather than tautological redefinitions or renamings of known patterns. This is the normal case of a method paper whose central ansatz draws on prior independent literature without load-bearing self-reference.
Axiom & Free-Parameter Ledger
Reference graph
Works this paper leans on
-
[1]
Build hybrid atomic orbitals (HAO) in sp, sp2, or sp3 hybridization and assign them to the vertices of the chemical graph
-
[2]
Align the orbitals with the direction of the bonds (edges)
-
[3]
Combine hybrid orbitals from connected vertices into bonding and anti-bonding types
-
[4]
Chemistry Localized Property-optimized Orbitals
Use this orbitals as initial guess in an orbital op- timizer For pure hydrogenic systems, this strategy can be im- plemented relatively straightforward (see for example [40]). For heterogenic molecules the manual prepara- tion of the initial guess is however tedious, which is why we resort to “Chemistry Localized Property-optimized Orbitals” (CLPO) [41] a...
-
[5]
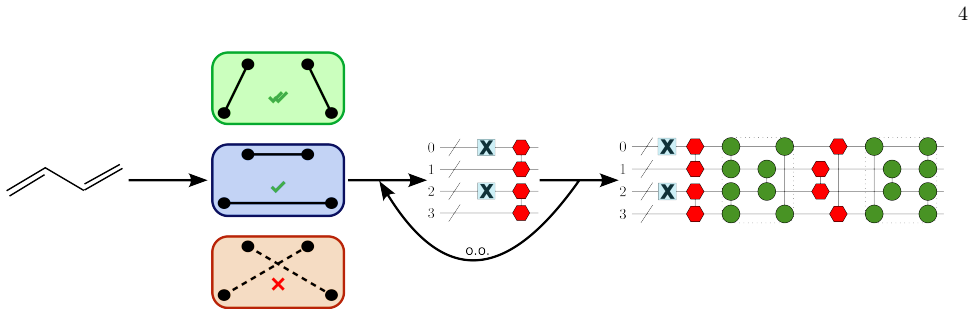
Con- struct a Lewis graph Based on the CLPO struc- ture 6
Initialize the CLPO through the blossom algo- rithm referenced in the previous section. Con- struct a Lewis graph Based on the CLPO struc- ture 6
-
[6]
(alternative) Select a Lewis graph for the molecule and manually configure the initial orbitals to re- semble the graph structure
-
[7]
Assign all CLPO to vertices in the graph
Decompose the graph into subgraphsC k ∈ G and select a representation (hardcore bosonic or fermionic). Assign all CLPO to vertices in the graph
-
[8]
Assemble SPA circuits for all hardcore bosonic parts
-
[9]
Assemble VQE circuit for the Fermionic parts
-
[10]
Compile the objective function (5) that represents the expectation value
-
[11]
Execute the VQE optimization (either classically, on quantum hardware, or mixed)
-
[12]
F/B R-DMRG
Optimize the molecular orbitals In this work we chose a multigraph schema, as illus- trated in Fig. 2 for the fermionic VQE parts in step 4. In the default construction illustrated above, all bosonic subspaces are classically simulable the underlying SPA circuit structure that restricts each bosonic subspace to a single quasi-particle. This limitation can...
-
[13]
A better spatial description of the wave function is required, usually addressed by increasing the basis set
Increasing the accuracy of the basis For most of the real-world relevant applications, a minimal basis such as STO-3G doesn’t provide accurate results. A better spatial description of the wave function is required, usually addressed by increasing the basis set. In quantum computational methods, this will lead to increased qubit requirements as well as res...
-
[14]
A.Aspuru-Guzik, A.D.Dutoi, P.J.Love,andM.Head- Gordon, Science309, 1704 (2005)
2005
-
[15]
P. J. Ollitrault, C. L. Cortes, J. F. Gonthier, R. M. Par- rish, D. Rocca, G.-L. Anselmetti, M. Degroote, N. Moll, R. Santagati, and M. Streif, Physical Review Letters 133, 250601 (2024)
2024
-
[16]
Fomichev, K
S. Fomichev, K. Hejazi, M. S. Zini, M. Kiser, J. Frax- anet, P.A.M.Casares, A.Delgado, J.Huh, A.-C.Voigt, J. E. Mueller, and J. M. Arrazola, PRX Quantum5, 040339 (2024)
2024
-
[17]
J. Lee, W. J. Huggins, M. Head-Gordon, and K. B. Whaley, Journal of Chemical Theory and Computation 15, 311 (2018)
2018
-
[18]
H. R. Grimsley, S. E. Economou, E. Barnes, and N. J. Mayhall, Nature Communications10, 1 (2019)
2019
-
[19]
H. G. A. Burton, D. Marti-Dafcik, D. P. Tew, and D. J. Wales, npj Quantum Information9, 75 (2023)
2023
-
[20]
H. G. A. Burton, Physical Review Research6, 023300 (2024)
2024
-
[21]
J. S. Kottmann and A. Aspuru-Guzik, Physical Re- view A105, 032449 (2022), arxiv:2105.03836 [physics, physics:quant-ph]
arXiv 2022
-
[22]
Ramôa, P
M. Ramôa, P. G. Anastasiou, L. P. Santos, N. J. May- hall, E. Barnes, and S. E. Economou, npj Quantum Information11, 86 (2025)
2025
-
[23]
C. J. Stein and M. Reiher, Journal of Chemical Theory and Computation12, 1760 (2016)
2016
-
[24]
L. Ding, S. Knecht, and C. Schilling, The Journal of Physical Chemistry Letters14, 11022 (2023)
2023
-
[25]
F. Tarocco, P. A. B. Haase, F. Pavošević, V. Kr- ishna, L. Guidoni, S. Knecht, and M. Stella, AEGISS – Atomic orbital and Entropy-based Guided Inference for Space Selection – A novel semi-automated active space selection workflow for quantum chemistry and quan- tum computing applications (2025), arXiv:2508.10671 [physics]
arXiv 2025
-
[26]
R. G. Shirazi, A. Zech, P. Pinski, and V. V. Ry- bkin, Performance of Automatic Active Space Se- lection for Electronic Excitation Energies (2025), arXiv:2511.05732 [physics.chem-ph]
arXiv 2025
-
[27]
J. J. Bao and D. G. Truhlar, Journal of Chemical The- ory and Computation15, 5308 (2019)
2019
-
[28]
F. J. de Arco Santos and J. S. Kottman, Quantum Science and Technology 10.1088/2058-9565/adbdee (2025)
-
[30]
J. S. Kottmann and F. Scala, Journal of Chemical The- ory and Computation20, 3514 (2024)
2024
-
[31]
J. S. Kottmann, P. Schleich, T. Tamayo-Mendoza, and A. Aspuru-Guzik, The Journal of Physical Chemistry Letters12, 663 (2021)
2021
-
[32]
P. Vaish and B. Rubenstein, Reducing the Cost of Unitary Coupled Cluster via Active Space Partitioning (2026), arXiv:2602.04783 [physics]
arXiv 2026
-
[33]
M. F. Lange and T. C. Berkelbach, Molecular physics 118, e1808726 (2020)
2020
-
[34]
Otten, M
M. Otten, M. R. Hermes, R. Pandharkar, Y. Alexeev, S. K. Gray, and L. Gagliardi, Journal of Chemical The- ory and Computation18, 7205 (2022)
2022
-
[35]
Q. Wang, R. D’Cunha, A. Mitra, S. K. Gray, M. Otten, and L. Gagliardi, The Journal of Physical Chemistry A 129, 7999 (2025)
2025
-
[36]
Yoshikawa, T
T. Yoshikawa, T. Takanashi, and H. Nakai, Journal of Chemical Theory and Computation18, 5360 (2022). 11
2022
-
[37]
Zhang, L
Y. Zhang, L. Cincio, C. F. A. Negre, P. Czarnik, P. J. Coles, P. M. Anisimov, S. M. Mniszewski, S. Tretiak, and P. A. Dub, npj Quantum Information8, 96 (2022)
2022
-
[38]
T. M. Henderson, I. W. Bulik, and G. E. Scuseria, The Journal of Chemical Physics142, 214116 (2015)
2015
-
[39]
T. M. Henderson, I. W. Bulik, T. Stein, and G. E. Scuseria, The Journal of Chemical Physics141, 244104 (2014)
2014
-
[40]
P. C. Hiberty, D. Danovich, and S. Shaik, inA Chemist’s Guide to Valence Bond Theory(John Wiley & Sons, Ltd, 2026) Chap. 4, pp. 83–95
2026
-
[41]
S. E. Ghasempouri, G. W. Dueck, and S. De Baerdemacker, The Journal of Physical Chem- istry A127, 8168 (2023)
2023
-
[42]
P. B. Karadakov, inChemical Modelling: Volume 5, edited by A. Hinchliffe (Royal Society of Chemistry, 2008)
2008
-
[43]
Simonetta, E
M. Simonetta, E. Gianinetti, and I. Vandoni, The Jour- nal of Chemical Physics48, 1579 (1968)
1968
-
[44]
Rumer, Göttinger Nachr.3, 337 (1932)
G. Rumer, Göttinger Nachr.3, 337 (1932)
1932
-
[45]
J. S. Kottmann, Quantum7, 1073 (2023)
2023
-
[46]
Granet and H
E. Granet and H. Dreyer, npj Quantum Information 10, 1 (2024)
2024
-
[47]
Google AI Quantum and Collaborators, Science369, 1084 (2020)
2020
-
[48]
Zhang, T
Z.-J. Zhang, T. H. Kyaw, J. Kottmann, M. Deg- roote, and A. Aspuru-Guzik, Quantum Sci. and Tech- nol. (2021)
2021
-
[49]
Bytautas, T
L. Bytautas, T. M. Henderson, C. A. Jiménez-Hoyos, J. K. Ellis, and G. E. Scuseria, The Journal of Chemical Physics135, 044119 (2011)
2011
-
[50]
P. A. Limacher, T. D. Kim, P. W. Ayers, P. A. Johnson, S. De Baerdemacker, D. Van Neck, and P. Bultinck, Molecular Physics112, 853 (2014)
2014
-
[51]
P. A. Limacher, The Journal of Chemical Physics164, 104117 (2026)
2026
-
[52]
W. J. Hehre, R. F. Stewart, and J. A. Pople, The Jour- nal of Chemical Physics51, 2657 (1969)
1969
-
[53]
Bincoletto, K
D. Bincoletto, K. Stein, J. Motyl, and J. S. Kottmann, Machine Learning: Science and Technology7, 035055 (2026)
2026
-
[54]
T. Y. Nikolaienko and L. A. Bulavin, International Journal of Quantum Chemistry119, e25798 (2019)
2019
-
[55]
Schollwoeck, Annals of Physics326, 96 (2011), arXiv:1008.3477 [cond-mat]
U. Schollwoeck, Annals of Physics326, 96 (2011), arXiv:1008.3477 [cond-mat]
Pith/arXiv arXiv 2011
-
[56]
G. K.-L. Chan and S. Sharma, Annual Review of Phys- ical Chemistry62, 465 (2011)
2011
-
[57]
A. Baiardi and M. Reiher, The Journal of Chemical Physics152, 040903 (2020), arXiv:1910.00137 [physics]
arXiv 2020
-
[58]
Yanai, Y
T. Yanai, Y. Kurashige, W. Mizukami, J. Chalupský, T. N. Lan, and M. Saitow, International Journal of Quantum Chemistry115, 283 (2015)
2015
-
[59]
A. O. Mitrushchenkov, G. Fano, R. Linguerri, and P. Palmieri, International Journal of Quantum Chem- istry112, 1606 (2012)
2012
-
[60]
F. Langkabel, S. Knecht, and J. S. Kottmann, arXiv preprint arXiv:2410.19116 (2024), arXiv:2410.19116 [quant-ph]
arXiv 2024
-
[61]
Langkabel, J
F. Langkabel, J. Kottmann, T. Scharfe, and T. Truong, FrayedEnds, github Repository
-
[62]
Crawford, B
O. Crawford, B. van Straaten, D. Wang, T. Parks, E. Campbell, and S. Brierley, Quantum5, 385 (2021)
2021
-
[63]
Z. P. Bansingh, T.-C. Yen, P. D. Johnson, and A. F. Izmaylov, The Journal of Physical Chemistry A126, 7007 (2022)
2022
-
[64]
Verteletskyi, T.-C
V. Verteletskyi, T.-C. Yen, and A. F. Izmaylov, The Journal of Chemical Physics152, 124114 (2020)
2020
-
[65]
Y. S. Yordanov, D. R. Arvidsson-Shukur, and C. H. Barnes, Physical Review A102, 062612 (2020)
2020
-
[66]
F. J. del Arco Santos, D. Bincoletto, J. S. Kottmann, and N. Roshani, Project-sunrise (2026), github Repos- itory
2026
-
[67]
J. S. Kottmann, S. Alperin-Lea, T. Tamayo-Mendoza, A. Cervera-Lierta, C. Lavigne, T.-C. Yen, V. Vertelet- skyi, P. Schleich, A. Anand, M. Degroote, S. Chaney, M. Kesibi, N. G. Curnow, B. Solo, G. Tsilimigkounakis, C. Zendejas-Morales, A. F. Izmaylov, and A. Aspuru- Guzik, Quantum Science and Technology6, 024009 (2021)
2021
-
[68]
Suzuki, Y
Y. Suzuki, Y. Kawase, Y. Masumura, Y. Hiraga, M. Nakadai, J. Chen, K. M. Nakanishi, K. Mi- tarai, R. Imai, S. Tamiya, T. Yamamoto, T. Yan, T. Kawakubo, Y. O. Nakagawa, Y. Ibe, Y. Zhang, H. Yamashita, H. Yoshimura, A. Hayashi, and K. Fujii, Quantum5, 559 (2021)
2021
-
[69]
McClean, N
J. McClean, N. Rubin, K. Sung, I. D. Kivlichan, X. Bonet-Monroig, Y. Cao, C. Dai, E. S. Fried, C. Gid- ney, B. Gimby,et al., Quantum Science and Technology (2020)
2020
-
[70]
Q. Sun, T. C. Berkelbach, N. S. Blunt, G. H. Booth, S. Guo, Z. Li, J. Liu, J. D. McClain, E. R. Sayfutyarova, S. Sharma, S. Wouters, and G. K.-L. Chan, Wiley In- terdisciplinary Review Compututational Molecular Sci- ence8, e1340 (2018)
2018
-
[71]
Q. Sun, X. Zhang, S. Banerjee, P. Bao, M. Barbry, N. S. Blunt, N. A. Bogdanov, G. H. Booth, J. Chen, Z.-H. Cui, J. J. Eriksen, andet. al., The Journal of Chemical Physics153, 024109 (2020)
2020
-
[72]
Q. Sun, X. Zhang, S. Banerjee, P. Bao, M. Barbry, N. S. Blunt, N. A. Bogdanov, G. H. Booth, J. Chen, Z.-H. Cui,et al.,TheJournalofChemicalPhysics153(2020)
2020
-
[73]
H. Zhai, H. R. Larsson, S. Lee, Z.-H. Cui, T. Zhu, C. Sun, L. Peng, R. Peng, K. Liao, J. Tölle, J. Yang, S. Li, and G. K.-L. Chan, The Journal of Chemical Physics159, 234801 (2023)
2023
-
[74]
J. S. Kottmann, A. Anand, and A. Aspuru-Guzik, Chemical Science12, 3497 (2021), arxiv:2011.05938 [physics, physics:quant-ph]
arXiv 2021
-
[75]
R. J. Harrison, G. Beylkin, F. A. Bischoff, J. A. Calvin, G. I. Fann, J. Fosso-Tande, D. Galindo, J. R. Ham- mond, R. Hartman-Baker, J. C. Hill,et al., SIAM Jour- nal on Scientific Computing38, S123 (2016)
2016
-
[76]
J. S. Kottmann, F. A. Bischoff, and E. F. Valeev, The Journal of Chemical Physics152, 074105 (2020)
2020
-
[77]
T. Y. Nikolaienko, L. A. Bulavin, and D. M. Hovorun, Computational and Theoretical Chemistry1050, 15 (2014). [65]qpicpackage: github.com/qpic (2026)
2014
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.