A general-purpose atomic cluster expansion interatomic potential for niobium
Pith reviewed 2026-07-02 10:32 UTC · model grok-4.3
The pith
An atomic cluster expansion potential for niobium achieves near-DFT accuracy for phonons, dislocations, and fracture simulations.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
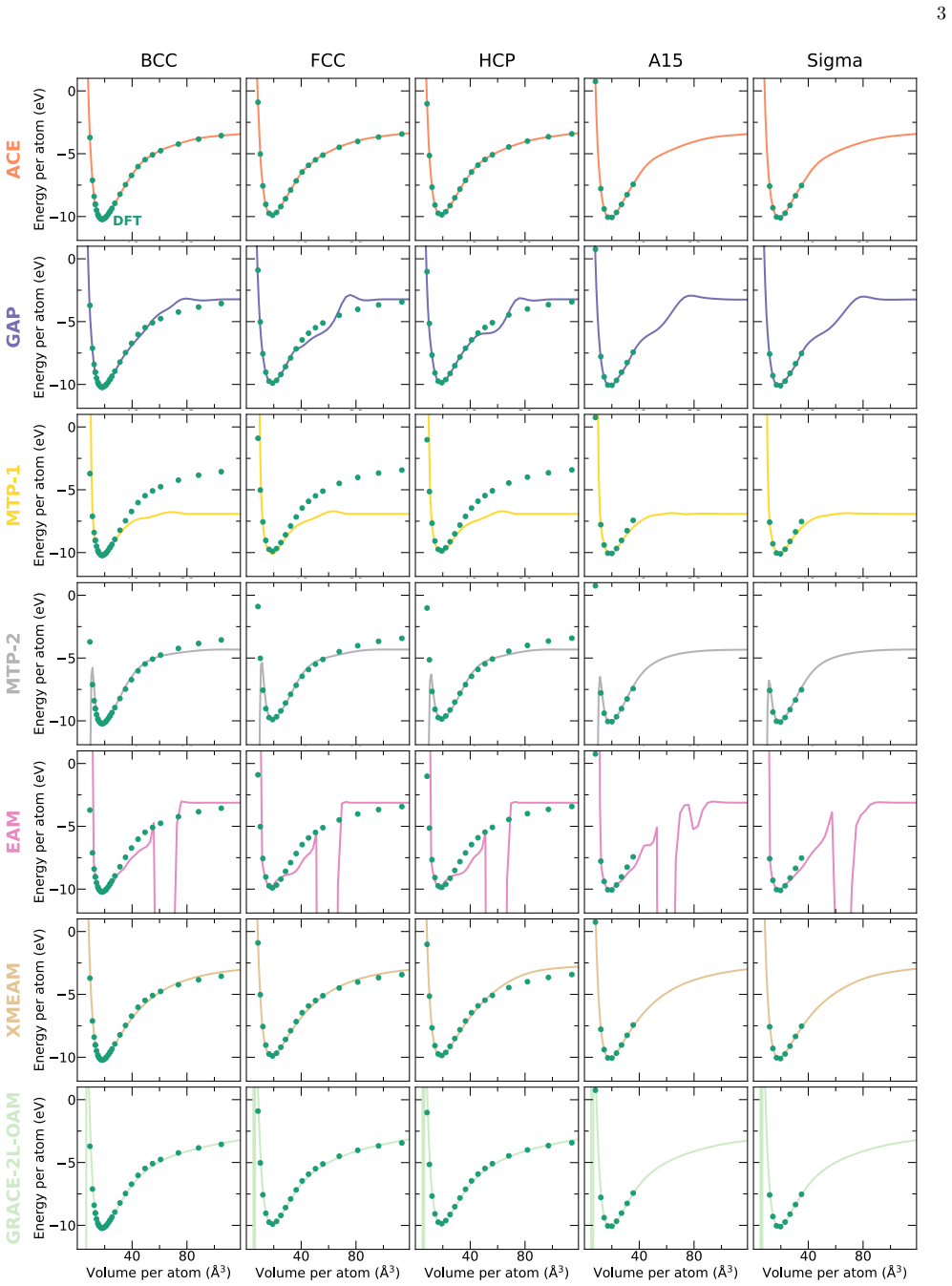
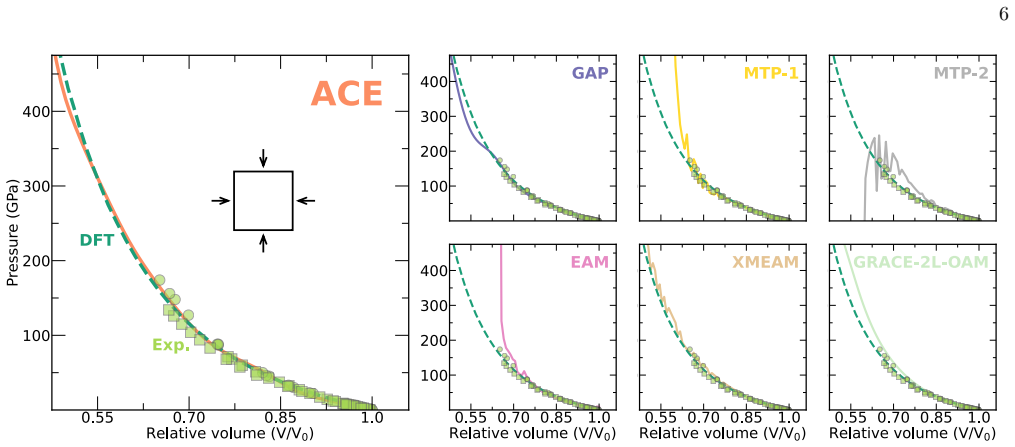
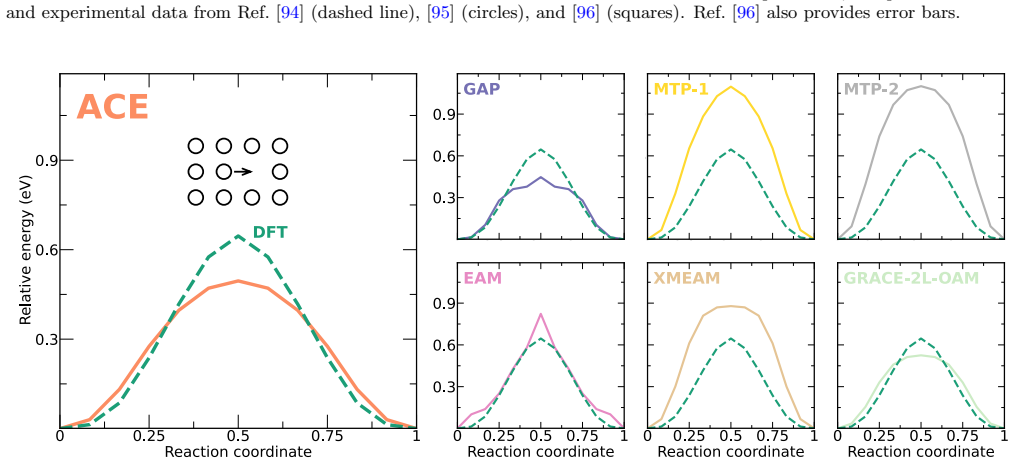
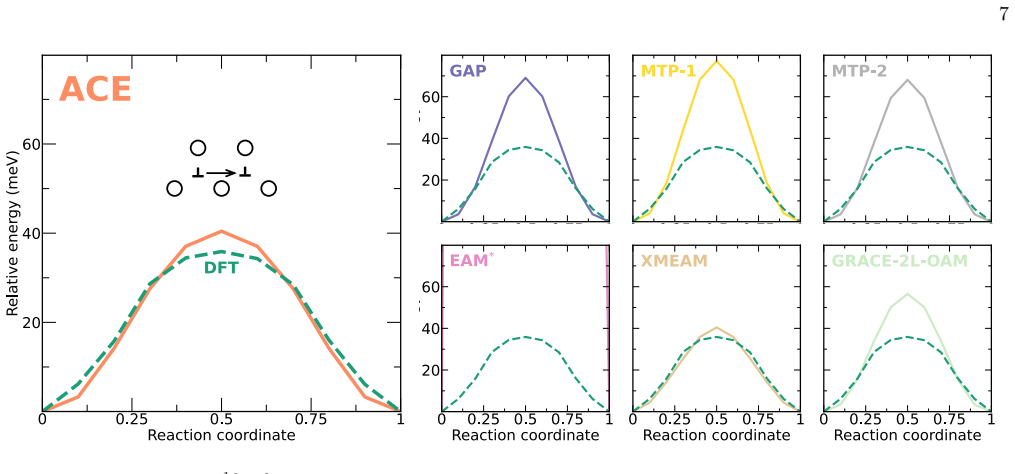
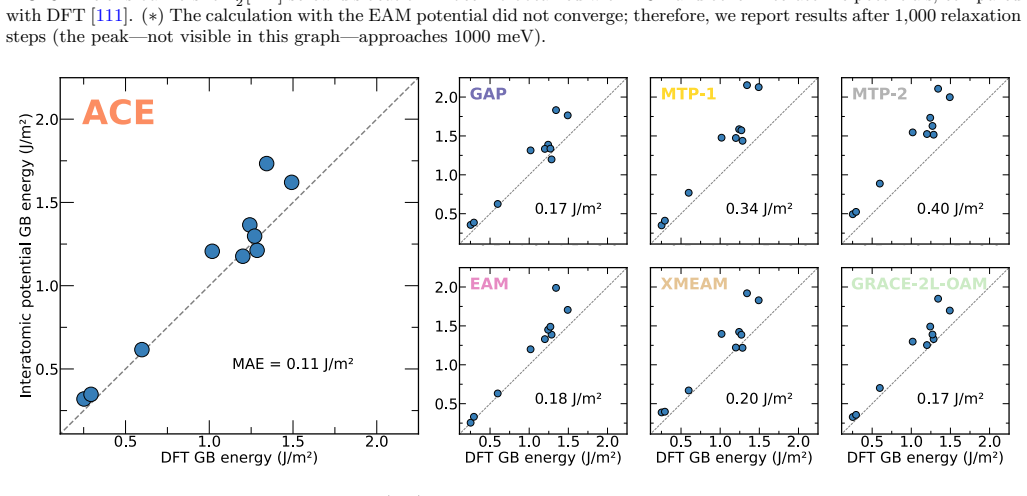
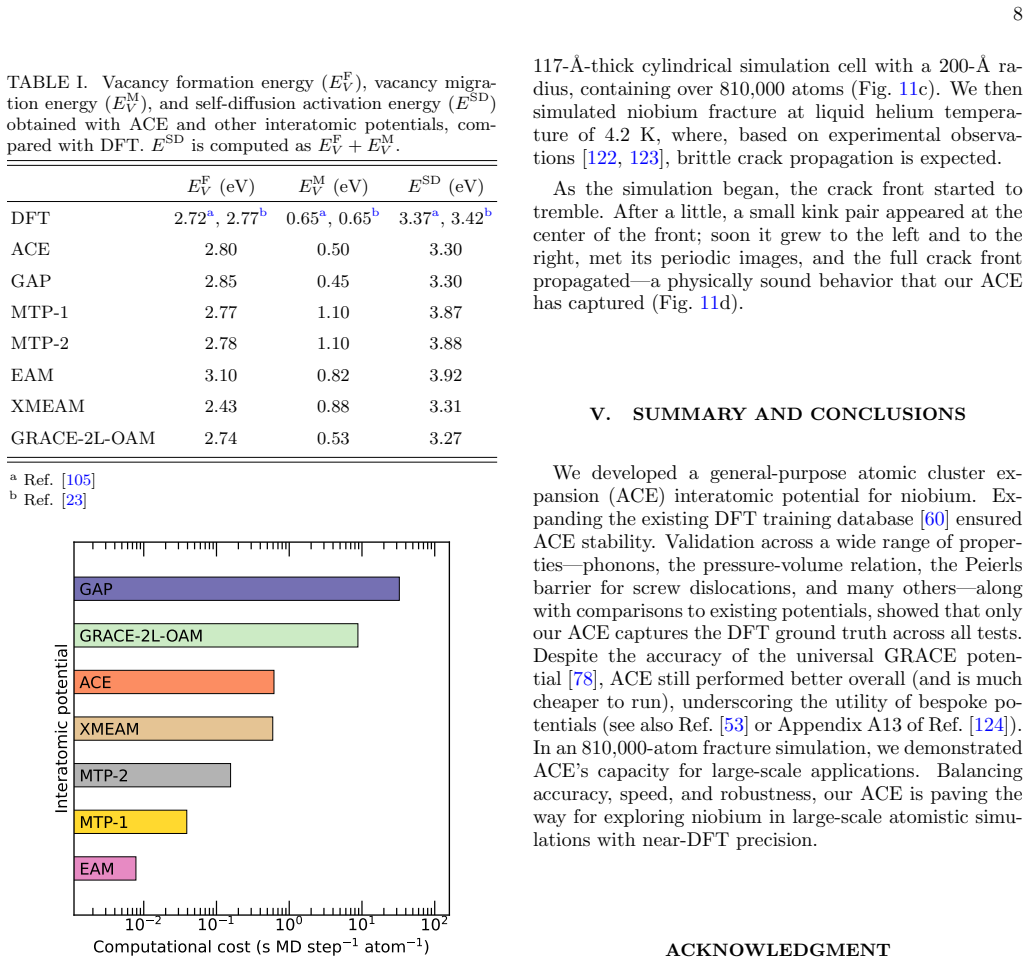
The authors construct an atomic cluster expansion interatomic potential for niobium. Trained on thousands of DFT structures spanning diverse local environments, the potential reproduces phonons, high-pressure behavior, energy barriers to dislocation glide and related properties at near-DFT precision. It balances accuracy, efficiency and robustness, and passes a stringent test by driving a near-million-atom molecular dynamics simulation of fracture.
What carries the argument
The atomic cluster expansion (ACE) interatomic potential, which expands atomic energies in a basis of cluster functions fitted directly to DFT reference data.
If this is right
- Large-scale molecular dynamics of niobium becomes feasible at near-DFT accuracy.
- Mechanical properties including fracture can be studied directly at million-atom scales.
- The potential serves as a reference for evaluating other interatomic models of niobium.
- Exploration of niobium under high strain or defect-rich conditions is now practical.
Where Pith is reading between the lines
- The same training strategy could produce reliable ACE potentials for other body-centered cubic transition metals.
- Accurate large-scale defect simulations may speed evaluation of niobium alloys for structural applications.
- If training-set diversity proves decisive, similar potentials could be built with fewer total DFT calculations.
Load-bearing premise
The thousands of DFT structures used for training span a sufficient diversity of local environments to generalize reliably to the tested properties and the fracture simulation.
What would settle it
A new DFT calculation for an untested property such as surface energy or stacking fault energy that deviates substantially from the ACE prediction, or visible mismatch between the million-atom fracture run and experimental crack behavior.
Figures

read the original abstract
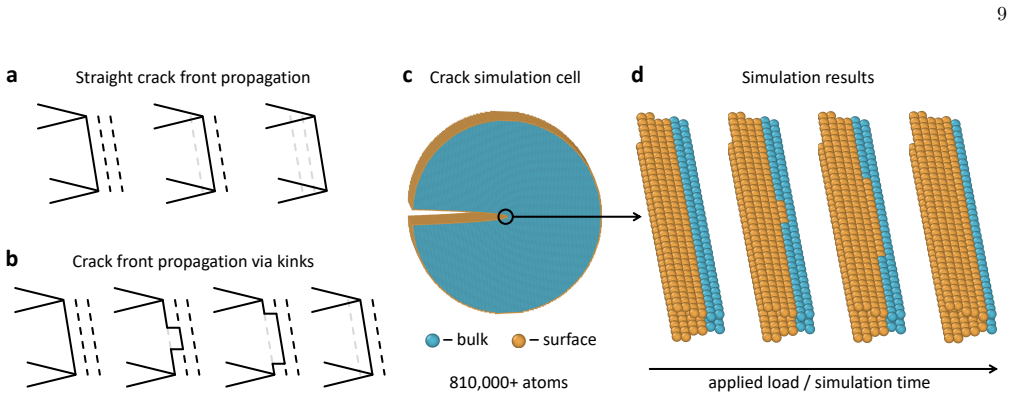
Niobium, a body-centered cubic transition metal, poses a challenge for interatomic potentials, which struggle to capture its properties, such as phonons, high-pressure behavior, energy barriers to dislocation glide, and others. To tackle this challenge, we constructed a general-purpose atomic cluster expansion (ACE) potential for niobium. We trained our ACE on thousands of density functional theory (DFT) structures spanning a diversity of local environments. We validated it across a range of properties and compared it with existing empirical and machine learning (ML) potentials, including a novel universal ML potential. The resulting ACE balances accuracy, efficiency, and robustness, enabling large-scale exploration of niobium with near-DFT precision. Finally, our ACE held its own in a stringent test: a near-million-atom molecular dynamics simulation of fracture
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript presents a general-purpose atomic cluster expansion (ACE) interatomic potential for niobium, trained on thousands of DFT structures spanning diverse local environments. It validates the potential against phonons, high-pressure behavior, dislocation glide energy barriers and other properties, compares it to empirical and ML potentials, and reports a near-million-atom MD fracture simulation as a stringent test, claiming the ACE achieves near-DFT precision while balancing accuracy, efficiency and robustness.
Significance. A well-validated general-purpose potential for Nb would enable reliable large-scale simulations of fracture and plasticity in a BCC transition metal where existing potentials have struggled; the scale of the fracture test is a notable strength if the training coverage is demonstrated to be adequate.
major comments (2)
- [Abstract and §2] Abstract and §2 (training data): the claim that the training set of 'thousands of DFT structures spanning a diversity of local environments' supports generalization to crack-tip and dislocation-core configurations in the fracture MD is not accompanied by quantitative coverage metrics (e.g., distribution of local strains, coordination numbers, or extrapolation grades), which is load-bearing for the central 'near-DFT precision' assertion in the million-atom test.
- [§4] §4 (fracture simulation): validation on phonons, high-pressure EOS and glide barriers does not automatically establish transferability to the high-strain, under-coordinated environments encountered during fracture; explicit tests (e.g., comparison of local atomic environments or error on held-out high-strain configurations) are required to substantiate the performance claim.
minor comments (2)
- [Abstract] Abstract: quantitative error metrics (RMSE, MAE) and error bars on the reported properties are absent, making it difficult to assess the 'near-DFT precision' claim without consulting the full results tables.
- [Methods] Notation: the definition of the ACE basis functions and the cutoff radius should be stated explicitly in the methods section for reproducibility.
Simulated Author's Rebuttal
We thank the referee for their constructive comments, which help clarify the presentation of our training data coverage and validation strategy. We respond to each major comment below and indicate revisions where appropriate.
read point-by-point responses
-
Referee: [Abstract and §2] Abstract and §2 (training data): the claim that the training set of 'thousands of DFT structures spanning a diversity of local environments' supports generalization to crack-tip and dislocation-core configurations in the fracture MD is not accompanied by quantitative coverage metrics (e.g., distribution of local strains, coordination numbers, or extrapolation grades), which is load-bearing for the central 'near-DFT precision' assertion in the million-atom test.
Authors: We agree that quantitative coverage metrics would strengthen the generalization argument. Section 2 describes the training set construction from diverse DFT configurations that include strained lattices, defects, and surfaces, but explicit distributions (e.g., of local strains or coordination numbers) or extrapolation-grade statistics are not reported. We will add these metrics in the revised manuscript to directly address coverage of crack-tip and dislocation-core environments. revision: yes
-
Referee: [§4] §4 (fracture simulation): validation on phonons, high-pressure EOS and glide barriers does not automatically establish transferability to the high-strain, under-coordinated environments encountered during fracture; explicit tests (e.g., comparison of local atomic environments or error on held-out high-strain configurations) are required to substantiate the performance claim.
Authors: We concur that property validations alone do not fully prove transferability to fracture environments. The manuscript relies on the scale and stability of the million-atom fracture simulation as a practical test of robustness. To strengthen this, we will add an analysis comparing local atomic environments sampled during the fracture run to the training distribution, along with any available error checks on additional high-strain test configurations, in the revised version. revision: yes
Circularity Check
No significant circularity in derivation chain
full rationale
The paper constructs an ACE potential by fitting to a large set of DFT reference structures and then validates performance on separate properties (phonons, high-pressure behavior, dislocation barriers) plus a large-scale MD demonstration. These steps follow standard supervised fitting plus external benchmarking; no equation or claim reduces by construction to its own inputs, no self-citation is load-bearing for a uniqueness result, and no ansatz is smuggled via prior work. The fracture simulation is an application test rather than a derived prediction forced by the training data.
Axiom & Free-Parameter Ledger
free parameters (1)
- ACE basis and coefficient parameters
axioms (1)
- domain assumption DFT calculations supply sufficiently accurate reference energies and forces for training an interatomic potential
Reference graph
Works this paper leans on
-
[1]
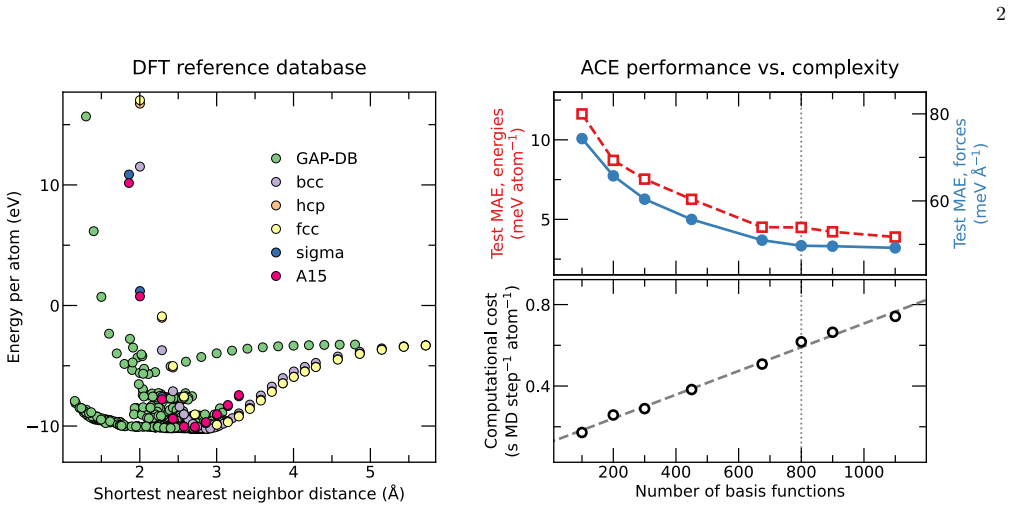
Density F unctional Theory (DFT) calculations DFT calculations were performed with settings iden- tical to those in the original GAP work [60], which pro- vided the data to train our ACE. We used theVASPpack- age [129–131] with the projector augmented-wave (PAW) method [132], the PBE generalized gradient approxima- tion (GGA-PBE) for exchange–correlation ...
-
[2]
Our nonlinear ACE employs the Finnis-Sinclair embed- ding
Atomic cluster expansion (ACE) training We trained our ACE using thepacemakerpackage [57]. Our nonlinear ACE employs the Finnis-Sinclair embed- ding. We selected a cutoff of 8 ˚A, determined through trial and error, to balance precision and speed. In train- ing, we assigned relative weights of 0.7 to energies and 0.3 to forces. This relatively high weight...
-
[3]
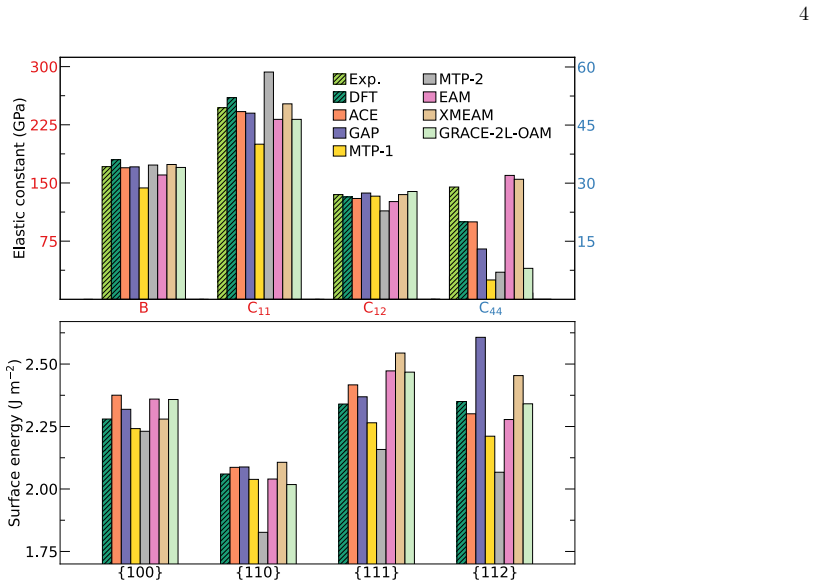
We computed the elastic constants, vacancy formation ener- gies, and phonons with theamstoolspackage [134]
V alidation details and setups For validation, we created a Python workflow based on the Atomic Simulation Environment (ASE) [127]. We computed the elastic constants, vacancy formation ener- gies, and phonons with theamstoolspackage [134]. The phonopypackage [135], integrated withinamstools, was used for phonon calculations. We used the nudged elas- tic b...
-
[4]
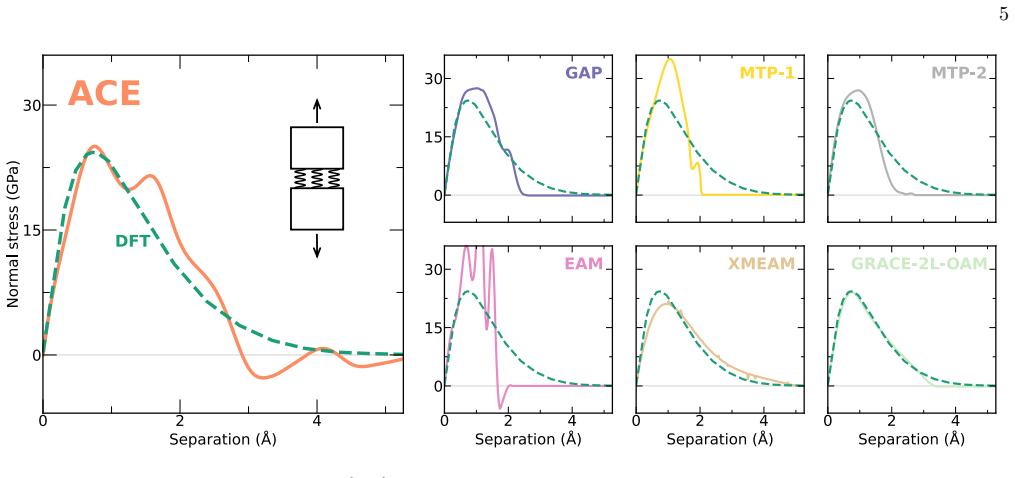
F racture simulations details and setups To model fracture, we employed a cylindrical simula- tion cell containing a pre-existing crack on one side, with the crack tip located at the center (Fig. 11c). Loading was applied via the mode-I stress intensity factor,K I, which uniquely determines the stress and strain fields around the crack tip [144]. For each...
-
[5]
Hatchett, Iii
C. Hatchett, Iii. an analysis of a mineral substance from north america, containing a metal bitberto unknown, Philos. Trans. R. Soc. London , 49 (1802)
-
[6]
G. De Marzi, Electronic band structure, lattice dy- namics, and related superconducting properties of nio- bium from first–principles calculations, High Perfor- mance Computing on CRESCO infrastructure: research activities and results 2015 , 99 (2016)
2015
-
[7]
C. M. Varma and W. Weber, Phonon dispersion in tran- sition metals, Phys. Rev. B19, 6142 (1979)
1979
-
[8]
M. W. Finnis, K. L. Kear, and D. G. Pettifor, Inter- atomic forces and phonon anomalies in bcc 3dtransition metals, Phys. Rev. Lett.52, 291 (1984)
1984
-
[9]
Landa, P
A. Landa, P. S¨ oderlind, O. I. Velikokhatnyi, I. I. Nau- mov, A. V. Ruban, O. E. Peil, and L. Vitos, Alloying- driven phase stability in group-VB transition metals un- der compression, Phys. Rev. B82, 144114 (2010)
2010
-
[10]
Liu and J
Z. Liu and J. Shang, First principles calculations of electronic properties and mechanical properties of bcc molybdenum and niobium, Rare Metals30, 354 (2011)
2011
-
[11]
Landa, P
A. Landa, P. S¨ oderlind, I. I. Naumov, J. E. Klepeis, and L. Vitos, Kohn anomaly and phase stability in group vb transition metals, Computation6(2018)
2018
-
[12]
Tidholm, O
J. Tidholm, O. Hellman, N. Shulumba, S. I. Simak, F. Tasn´ adi, and I. A. Abrikosov, Temperature depen- dence of the Kohn anomaly in bcc Nb from first- principles self-consistent phonon calculations, Phys. Rev. B101, 115119 (2020)
2020
-
[13]
Errandonea, L
D. Errandonea, L. Burakovsky, D. L. Preston, S. G. MacLeod, D. Santamar´ ıa-Perez, S. Chen, H. Cynn, S. I. Simak, M. I. McMahon, J. E. Proctor, and M. Mezouar, Experimental and theoretical confirmation of an or- thorhombic phase transition in niobium at high pressure and temperature, Commun. Mater.1, 60 (2020)
2020
-
[14]
Hohenberg and W
P. Hohenberg and W. Kohn, Inhomogeneous electron gas, Phys. Rev.136, B864 (1964)
1964
-
[15]
Kohn and L
W. Kohn and L. J. Sham, Self-consistent equations in- cluding exchange and correlation effects, Phys. Rev. 140, A1133 (1965)
1965
-
[16]
Lejaeghereet al., Reproducibility in density func- tional theory calculations of solids, Science351, aad3000 (2016)
K. Lejaeghereet al., Reproducibility in density func- tional theory calculations of solids, Science351, aad3000 (2016)
2016
-
[17]
M. S. Daw and M. I. Baskes, Embedded-atom method: Derivation and application to impurities, surfaces, and other defects in metals, Phys. Rev. B29, 6443 (1984)
1984
-
[18]
Rodney, Atomic modeling of irradiation-induced hardening, C
D. Rodney, Atomic modeling of irradiation-induced hardening, C. R. Phys.9, 418 (2008), Materials sub- jected to fast neutron irradiation
2008
-
[19]
Zhang, J
R. Zhang, J. Wang, I. Beyerlein, and T. Germann, Twinning in bcc metals under shock loading: a chal- lenge to empirical potentials, Philos. Mag. Lett.91, 731 (2011)
2011
-
[20]
J. J. M¨ oller and E. Bitzek, Comparative study of embed- ded atom potentials for atomistic simulations of fracture inα-iron, Model. Simul. Mater. Sci. Eng.22, 045002 (2014)
2014
-
[21]
Bitzek, J
E. Bitzek, J. R. Kermode, and P. Gumbsch, Atomistic aspects of fracture, Int. J. Fract.191, 13 (2015)
2015
-
[22]
Hiremath, S
P. Hiremath, S. Melin, E. Bitzek, and P. A. T. Olsson, Effects of interatomic potential on fracture behaviour in single- and bicrystalline tungsten, Comput. Mater. Sci. 207, 111283 (2022)
2022
-
[23]
Allera, T
A. Allera, T. D. Swinburne, A. M. Goryaeva, B. Bi- envenu, F. Ribeiro, M. Perez, M.-C. Marinica, and D. Rodney, Activation entropy of dislocation glide in body-centered cubic metals from atomistic simulations, 11 Nat. Commun.16, 8367 (2025)
2025
-
[24]
Jacobset al., A practical guide to machine learning interatomic potentials – status and future, Curr
R. Jacobset al., A practical guide to machine learning interatomic potentials – status and future, Curr. Opin. Solid State Mater. Sci.35, 101214 (2025)
2025
-
[25]
Morrow, J
J. Morrow, J. Gardner, and V. Deringer, How to vali- date machine-learned interatomic potentials, J. Chem. Phys.158, 121501 (2023)
2023
-
[26]
Behler and M
J. Behler and M. Parrinello, Generalized neural-network representation of high-dimensional potential-energy sur- faces, Phys. Rev. Lett.98, 146401 (2007)
2007
-
[27]
A. P. Bart´ ok, M. C. Payne, R. Kondor, and G. Cs´ anyi, Gaussian approximation potentials: The accuracy of quantum mechanics, without the electrons, Phys. Rev. Lett.104, 136403 (2010)
2010
-
[28]
Thompson, L
A. Thompson, L. Swiler, C. Trott, S. Foiles, and G. Tucker, Spectral neighbor analysis method for auto- mated generation of quantum-accurate interatomic po- tentials, J. Comput. Phys.285, 316 (2015)
2015
-
[29]
A. V. Shapeev, Moment tensor potentials: A class of systematically improvable interatomic potentials, Mul- tiscale Model. Simul.14, 1153 (2016)
2016
-
[30]
Drautz, Atomic cluster expansion for accurate and transferable interatomic potentials, Phys
R. Drautz, Atomic cluster expansion for accurate and transferable interatomic potentials, Phys. Rev. B99, 014104 (2019)
2019
-
[31]
C. J. Pickard, Ephemeral data derived potentials for random structure search, Phys. Rev. B106, 014102 (2022)
2022
-
[32]
Batatia, D
I. Batatia, D. P. Kovacs, G. Simm, C. Ortner, and G. Csanyi, Mace: Higher order equivariant message passing neural networks for fast and accurate force fields, inAdv. Neural Inf. Process. Syst., Vol. 35, edited by S. Koyejo, S. Mohamed, A. Agarwal, D. Belgrave, K. Cho, and A. Oh (Curran Associates, Inc., 2022) pp. 11423–11436
2022
-
[33]
Batzner, A
S. Batzner, A. Musaelian, L. Sun, M. Geiger, J. P. Mailoa, M. Kornbluth, N. Molinari, T. E. Smidt, and B. Kozinsky, E(3)-equivariant graph neural networks for data-efficient and accurate interatomic potentials, Nat. Commun.13, 2453 (2022)
2022
-
[34]
Cheng, Cartesian atomic cluster expansion for ma- chine learning interatomic potentials, npj Comput
B. Cheng, Cartesian atomic cluster expansion for ma- chine learning interatomic potentials, npj Comput. Mater.10, 157 (2024)
2024
-
[35]
D. J. Burrill, C. Liu, M. G. Taylor, M. J. Cawkwell, D. Perez, E. R. Batista, N. Lubbers, and P. Yang, Mltb: Enhancing transferability and extensibility of density functional tight-binding theory with many-body inter- action corrections, J. Chem. Theory Comput21, 1089 (2025)
2025
-
[36]
Jinnouchi, J
R. Jinnouchi, J. Lahnsteiner, F. Karsai, G. Kresse, and M. Bokdam, Phase transitions of hybrid perovskites simulated by machine-learning force fields trained on the fly with bayesian inference, Phys. Rev. Lett.122, 225701 (2019)
2019
-
[37]
Kostiuchenko, F
T. Kostiuchenko, F. K¨ ormann, J. Neugebauer, and A. Shapeev, Impact of lattice relaxations on phase tran- sitions in a high-entropy alloy studied by machine- learning potentials, npj Comput. Mater.5, 55 (2019)
2019
-
[38]
Cheng, E
B. Cheng, E. A. Engel, J. Behler, C. Dellago, and M. Ce- riotti, Ab initio thermodynamics of liquid and solid wa- ter, Proc. Natl Acad. Sci. USA116, 1110 (2019)
2019
-
[39]
Cheng, G
B. Cheng, G. Mazzola, C. J. Pickard, and M. Ceriotti, Evidence for supercritical behaviour of high-pressure liquid hydrogen, Nature585, 217 (2020)
2020
-
[40]
V. L. Deringer, C. J. Pickard, and G. Cs´ anyi, Data- driven learning of total and local energies in elemental boron, Phys. Rev. Lett.120, 156001 (2018)
2018
-
[41]
Timmermann, F
J. Timmermann, F. Kraushofer, N. Resch, P. Li, Y. Wang, Z. Mao, M. Riva, Y. Lee, C. Staacke, M. Schmid, C. Scheurer, G. S. Parkinson, U. Diebold, and K. Reuter, IrO 2 Surface Complexions Identified through Machine Learning and Surface Investigations, Phys. Rev. Lett.125, 206101 (2020)
2020
-
[42]
E. V. Podryabinkin, E. V. Tikhonov, A. V. Shapeev, and A. R. Oganov, Accelerating crystal structure pre- diction by machine-learning interatomic potentials with active learning, Phys. Rev. B99, 064114 (2019)
2019
-
[43]
J. R. Cendagorta, H. Shen, Z. Baˇ ci´ c, and M. E. Tuck- erman, Enhanced sampling path integral methods us- ing neural network potential energy surfaces with appli- cation to diffusion in hydrogen hydrates, Adv. Theory Simul.4, 2000258 (2021)
2021
-
[44]
V. L. Deringer, N. Bernstein, G. Cs´ anyi, C. Ben Mah- moud, M. Ceriotti, M. Wilson, D. A. Drabold, and S. R. Elliott, Origins of structural and electronic transitions in disordered silicon, Nature589, 59 (2021)
2021
-
[45]
Lopanitsyna, G
N. Lopanitsyna, G. Fraux, M. A. Springer, S. De, and M. Ceriotti, Modeling high-entropy transition metal al- loys with alchemical compression, Phys. Rev. Mater.7, 045802 (2023)
2023
-
[46]
Kapil, C
V. Kapil, C. Schran, A. Zen, J. Chen, C. J. Pickard, and A. Michaelides, The first-principles phase diagram of monolayer nanoconfined water, Nature609, 512 (2022)
2022
-
[47]
Zhang, G
L. Zhang, G. Cs´ anyi, E. Van Der Giessen, and F. Maresca, Atomistic fracture in bcc iron revealed by active learning of gaussian approximation potential, npj Comput. Mater.9, 217 (2023)
2023
-
[48]
J. Lan, A. Palizhati, M. Shuaibi, B. M. Wood, B. Wan- der, A. Das, M. Uyttendaele, C. L. Zitnick, and Z. W. Ulissi, AdsorbML: a leap in efficiency for adsorption en- ergy calculations using generalizable machine learning potentials, npj Comput. Mater.9, 172 (2023)
2023
-
[49]
Y. Zhou, W. Zhang, E. Ma, and V. L. Deringer, Device- scale atomistic modelling of phase-change memory ma- terials, Nat. Electron.6, 746 (2023)
2023
-
[50]
Y. Zhou, D. F. Thomas du Toit, S. R. Elliott, W. Zhang, and V. L. Deringer, Full-cycle device-scale simulations of memory materials with a tailored atomic-cluster- expansion potential, Nat. Commun.16, 8688 (2025)
2025
-
[51]
Sheriff, Y
K. Sheriff, Y. Cao, T. Smidt, and R. Freitas, Quantify- ing chemical short-range order in metallic alloys, Proc. Natl Acad. Sci. USA121, e2322962121 (2024)
2024
-
[52]
X. Xu, X. Zhang, E. Bitzek, S. Schmauder, and B. Grabowski, Origin of the yield stress anomaly in L12 intermetallics unveiled with physically informed machine-learning potentials, Acta Mater.281, 120423 (2024)
2024
-
[53]
X. Wang, S. Xu, W.-R. Jian, X.-G. Li, Y. Su, and I. J. Beyerlein, Generalized stacking fault energies and peierls stresses in refractory body-centered cubic met- als from machine learning-based interatomic potentials, Comput. Mater. Sci.192, 110364 (2021)
2021
-
[54]
Ngoipala, C
A. Ngoipala, C. Schott, V. Briega-Martos, M. Qamar, M. Mrovec, S. Javan Nikkhah, T. O. Schmidt, L. Dev- ille, A. Capogrosso, L. Moumaneix,et al., Hydride- induced reconstruction of pd electrode surfaces: A combined computational and experimental study, Adv. Mater.37, 2410951 (2025)
2025
-
[55]
Zhang, S
X. Zhang, S. V. Divinski, and B. Grabowski, Ab initio 12 machine-learning unveils strong anharmonicity in non- arrhenius self-diffusion of tungsten, Nat. Commun.16, 394 (2025)
2025
-
[56]
M. O. Sauer, P. M. Lyngby, and K. S. Thygesen, Dispersion-corrected machine learning potentials for 2D van der Waals materials, Phys. Rev. Mater.9, 074007 (2025)
2025
-
[57]
L. J. Conway and C. J. Pickard, Accelerating crys- tal structure prediction using data-derived potentials: High-pressure binary hydrides, Ann. Phys.538, e00608 (2026)
2026
-
[58]
A. Allera, L. Ventelon, M.-C. Marinica, D. Rodney, and L. Proville, Revisiting quantum effects on dislocation glide in bcc metals from dft calculations and machine- learning potentials (2026), arXiv:2606.17954
work page internal anchor Pith review Pith/arXiv arXiv 2026
-
[59]
Mishin, Machine-learning interatomic potentials for materials science, Acta Mater.214, 116980 (2021)
Y. Mishin, Machine-learning interatomic potentials for materials science, Acta Mater.214, 116980 (2021)
2021
-
[60]
Zhanget al., Roadmap for the development of machine learning-based interatomic potentials, Model
Y.-W. Zhanget al., Roadmap for the development of machine learning-based interatomic potentials, Model. Simul. Mater. Sci. Eng.33, 023301 (2025)
2025
-
[61]
Lysogorskiy, C
Y. Lysogorskiy, C. van der Oord, A. Bochkarev, S. Menon, M. Rinaldi, T. Hammerschmidt, M. Mrovec, A. Thompson, G. Cs´ anyi, C. Ortner, and R. Drautz, Performant implementation of the atomic cluster expan- sion (PACE) and application to copper and silicon, npj Comput. Mater.7, 97 (2021)
2021
-
[62]
Zhang, G
L. Zhang, G. Cs´ anyi, E. van der Giessen, and F. Maresca, Efficiency, accuracy, and transferability of machine learning potentials: Application to dislocations and cracks in iron, Acta Mater.270, 119788 (2024)
2024
-
[63]
Leimeroth, L
N. Leimeroth, L. C. Erhard, K. Albe, and J. Rohrer, Machine-learning interatomic potentials from a users perspective: a comparison of accuracy, speed and data efficiency, Model. Simul. Mater. Sci. Eng.33, 065012 (2025)
2025
-
[64]
Byggm¨ astar, K
J. Byggm¨ astar, K. Nordlund, and F. Djurabekova, Gaussian approximation potentials for body-centered- cubic transition metals, Phys. Rev. Mater.4, 093802 (2020)
2020
-
[65]
Ben Mahmoud, J
C. Ben Mahmoud, J. Gardner, and V. Deringer, Data as the next challenge in atomistic machine learning, Nat. Comput. Sci.4, 384 (2024)
2024
-
[66]
Byggm¨ astaret al., GAP DFT reference database for niobium,https://gitlab.com/acclab/gap-data/ -/tree/master/Nb/training-data(2019)
2019
-
[67]
R. Wang, L. Zhu, S. Pattamatta, D. J. Srolovitz, and Z. Wu, The taming of the screw: Dislocation cores in BCC metals and alloys, Mater. Today (2024)
2024
-
[68]
Kunzmann, T
S. Kunzmann, T. Hammerschmidt, G. Schierning, and A. Gr¨ unebohm, Ab initio study of transition paths be- tween (meta)stable phases of Nb and Ta-substituted Nb, Phys. Rev. Mater.8, 033603 (2024)
2024
-
[69]
Bochkarev, Y
A. Bochkarev, Y. Lysogorskiy, S. Menon, M. Qamar, M. Mrovec, and R. Drautz, Efficient parametrization of the atomic cluster expansion,Phys. Rev. Mater.6, 013804 (2022)
2022
-
[70]
D. P. Kov´ acs, C. van der Oord, J. Kucera, A. E. A. Allen, D. J. Cole, C. Ortner, and G. Cs´ anyi, Lin- ear atomic cluster expansion force fields for organic molecules: Beyond RMSE, J. Chem. Theory Comput. 17, 7696 (2021)
2021
- [71]
-
[72]
S. Yin, Y. Zuo, A. Abu-Odeh, H. Zheng, X.-G. Li, J. Ding, S. P. Ong, M. Asta, and R. O. Ritchie, Atom- istic simulations of dislocation mobility in refractory high-entropy alloys and the effect of chemical short- range order, Nat. Commun.12, 4873 (2021)
2021
-
[73]
J. H. Jung, P. Srinivasan, A. Forslund, and B. Grabowski, High-accuracy thermodynamic proper- ties to the melting point from ab initio calculations aided by machine-learning potentials, npj Comput. Mater.9, 3 (2023)
2023
-
[74]
The authors of the MTP-2 developed one more poten- tial for niobium. For benchmarking, we considered their original potential, as the latest was explicitly designed for a narrow application addressing dislocations and its training was limited to a few hundred structures related to dislocations
-
[75]
M. R. Fellinger, H. Park, and J. W. Wilkins, Force- matched embedded-atom method potential for niobium, Phys. Rev. B81, 144119 (2010)
2010
-
[76]
Wang,Developing Extended Modified Embedded- Atom Method Potentials for Atomistic Modelling on Plasticity and Fracture Behaviours of Metals, Ph.D
R. Wang,Developing Extended Modified Embedded- Atom Method Potentials for Atomistic Modelling on Plasticity and Fracture Behaviours of Metals, Ph.D. the- sis, City University of Hong Kong (2023)
2023
-
[77]
R. Wang, X. Ma, L. Zhang, H. Wang, D. J. Srolovitz, T. Wen, and Z. Wu, Classical and machine learning interatomic potentials for bcc vanadium, Phys. Rev. Mater.6, 113603 (2022)
2022
-
[78]
A. Loew, D. Sun, H.-C. Wang, S. Botti, and M. A. L. Marques, Universal machine learning interatomic poten- tials are ready for phonons, npj Comput. Mater.11, 178 (2025)
2025
-
[79]
Shuang, Z
F. Shuang, Z. Wei, K. Liu, W. Gao, and P. Dey, Uni- versal machine learning interatomic potentials poised to supplant DFT in modeling general defects in metals and random alloys, Mach. Learn. Sci. Tech.6, 030501 (2025)
2025
-
[80]
Sharma, A
K. Sharma, A. Loew, H. Wang, F. A. Nilsson, M. Jain, M. A. L. Marques, and K. S. Thygesen, Accelerating point defect photo-emission calculations with machine learning interatomic potentials, npj Comput. Mater.11, 334 (2025)
2025
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.