Predicting Novel Stable Materials for Experimental Synthesis
Pith reviewed 2026-07-03 10:15 UTC · model grok-4.3
The pith
A hierarchical screening protocol narrows 894 computationally stable materials to 25 high-confidence targets for experimental synthesis.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
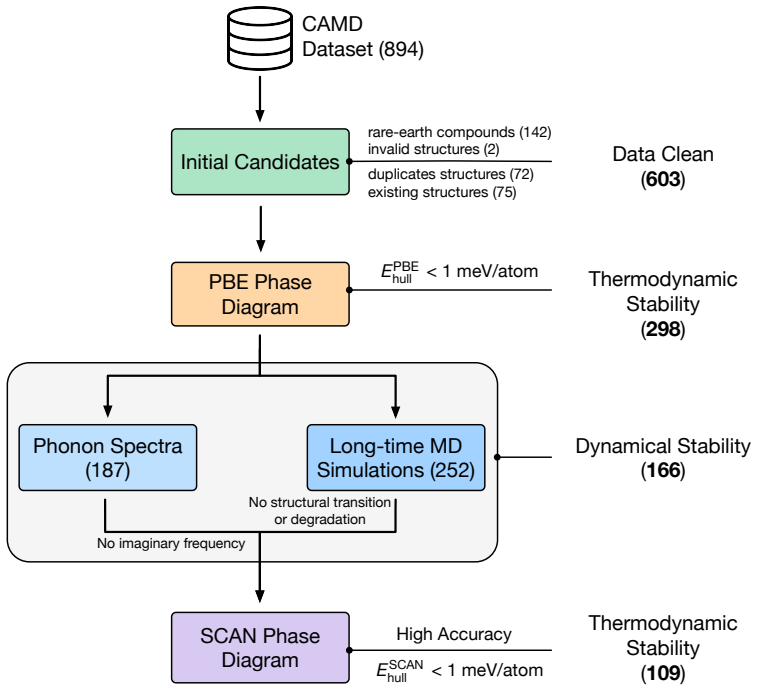
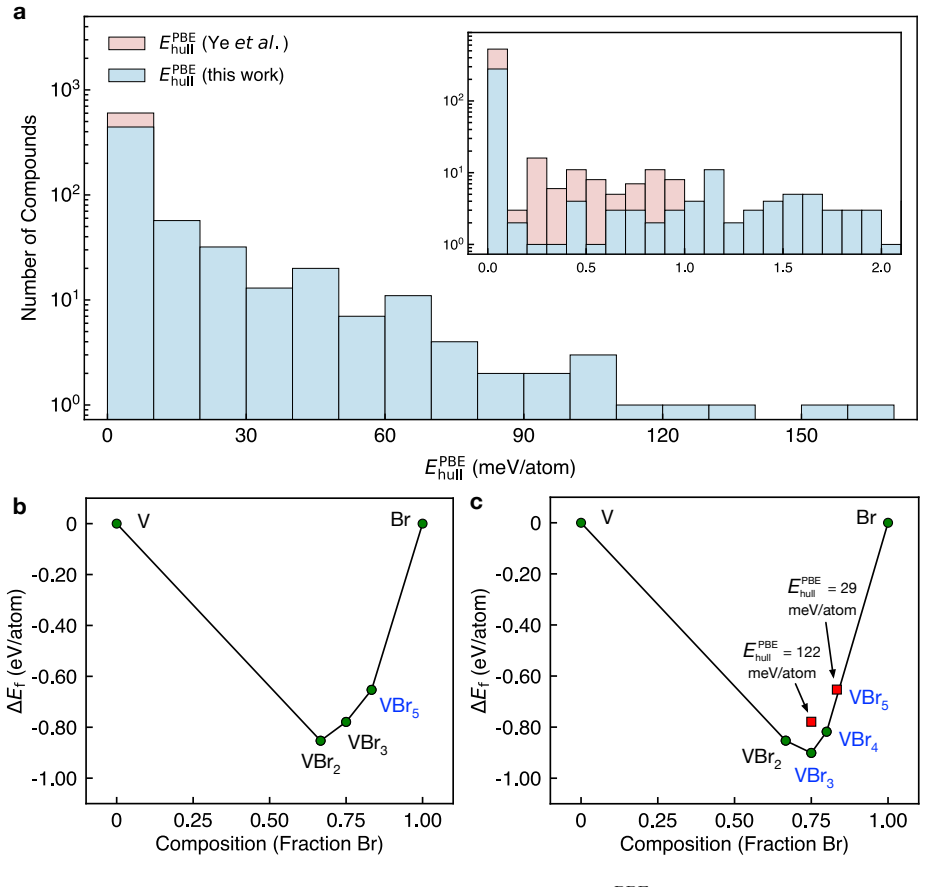
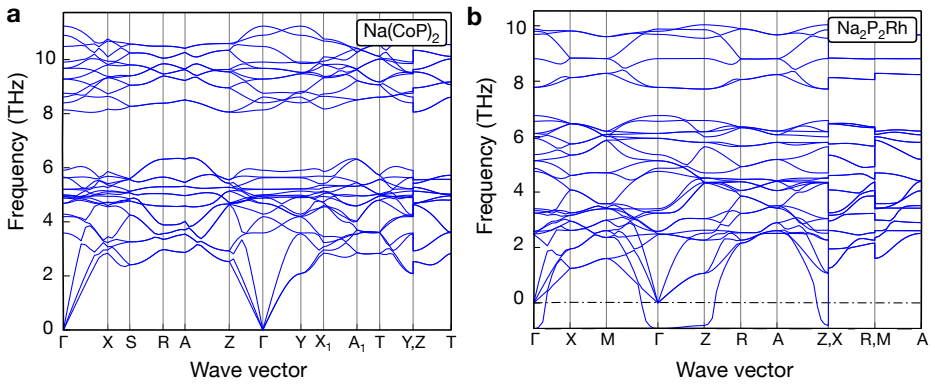
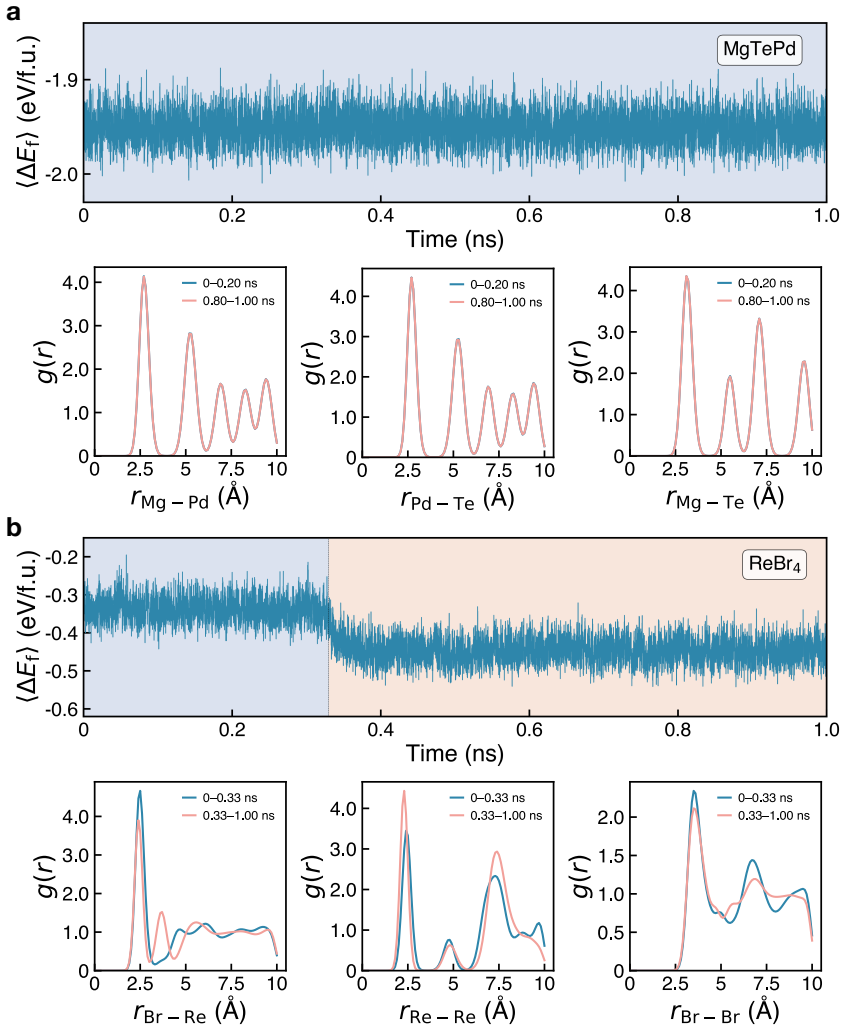
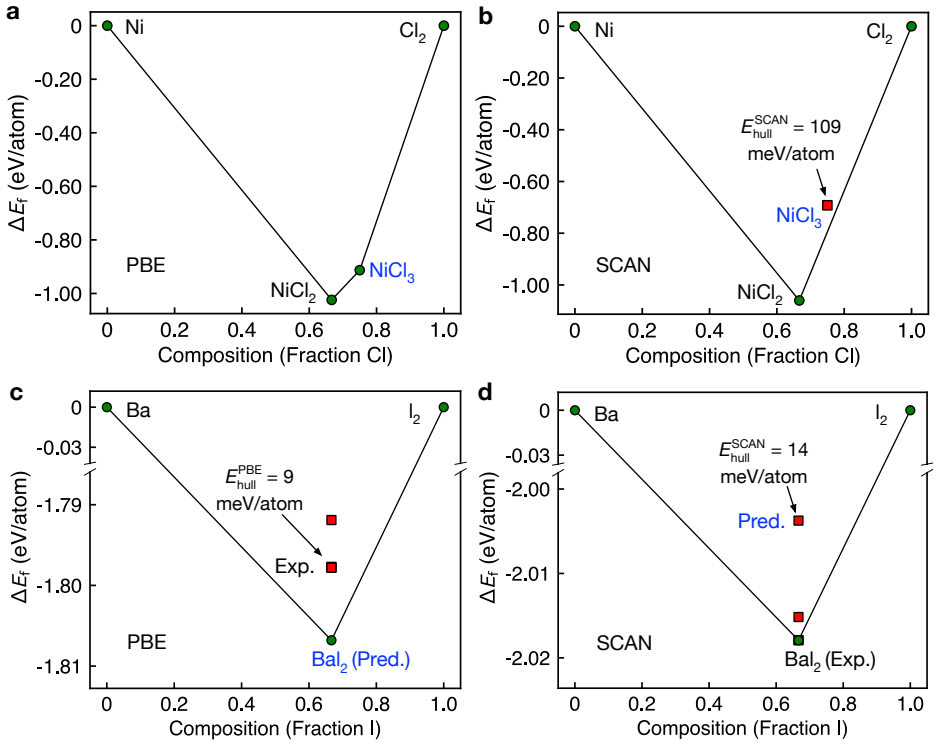
The central claim is that applying the hierarchical protocol to the 894 stable materials previously reported in Sci. Data 9, 302 (2022) yields 25 high-confidence targets for experimental synthesis after successive PBE, dynamical, and SCAN filters. The workflow first curates 603 unique structures, of which only 298 remain thermodynamically stable on the complete PBE phase diagrams. Dynamical screening then identifies 166 materials stable under both harmonic-phonon and finite-temperature molecular dynamics criteria, SCAN phase diagrams further narrow the set to 109, and the final combination of decomposition enthalpy with chemical-space completeness selects the 25 candidates.
What carries the argument
The hierarchical screening framework that combines PBE-based thermodynamic stability on complete phase diagrams, dynamical-stability screening enabled by universal machine-learning interatomic potentials, and SCAN-based thermodynamic refinement.
If this is right
- Competing phases must be considered, since only 298 of the 603 unique structures remain thermodynamically stable on full PBE phase diagrams.
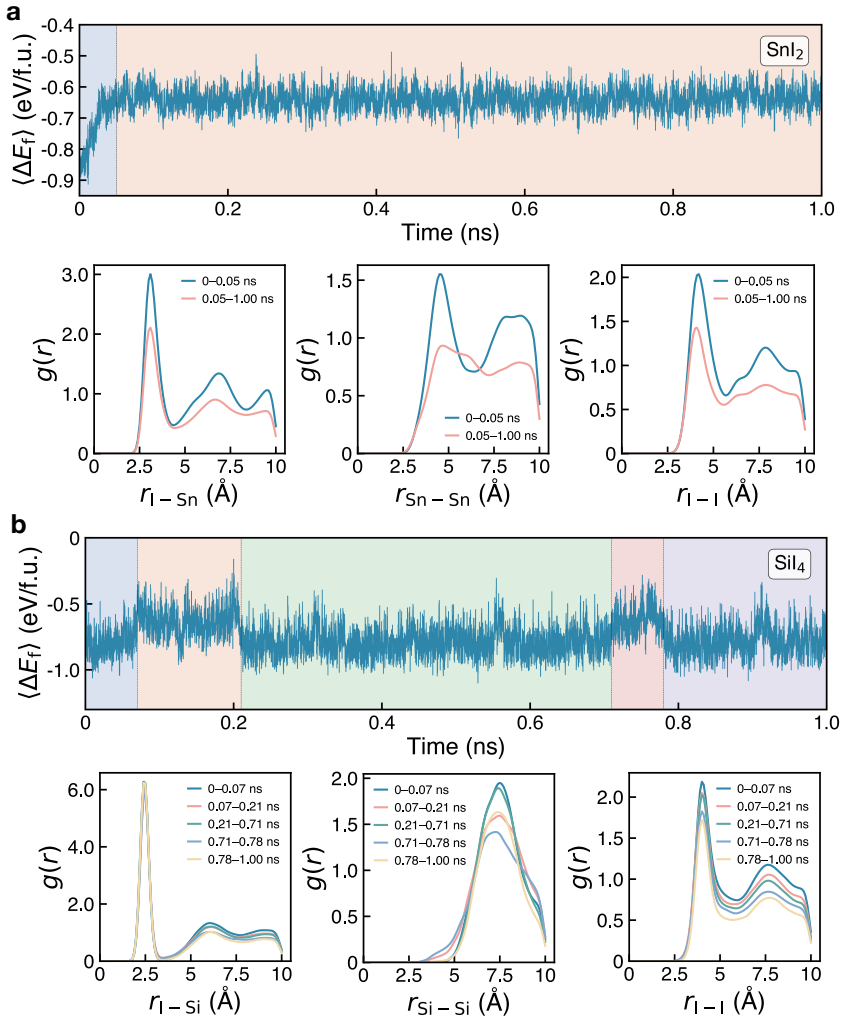
- Dynamical screening with machine-learning potentials reduces the candidate pool to 166 materials that satisfy both phonon and molecular-dynamics criteria.
- SCAN refinement further narrows the list to 109 materials.
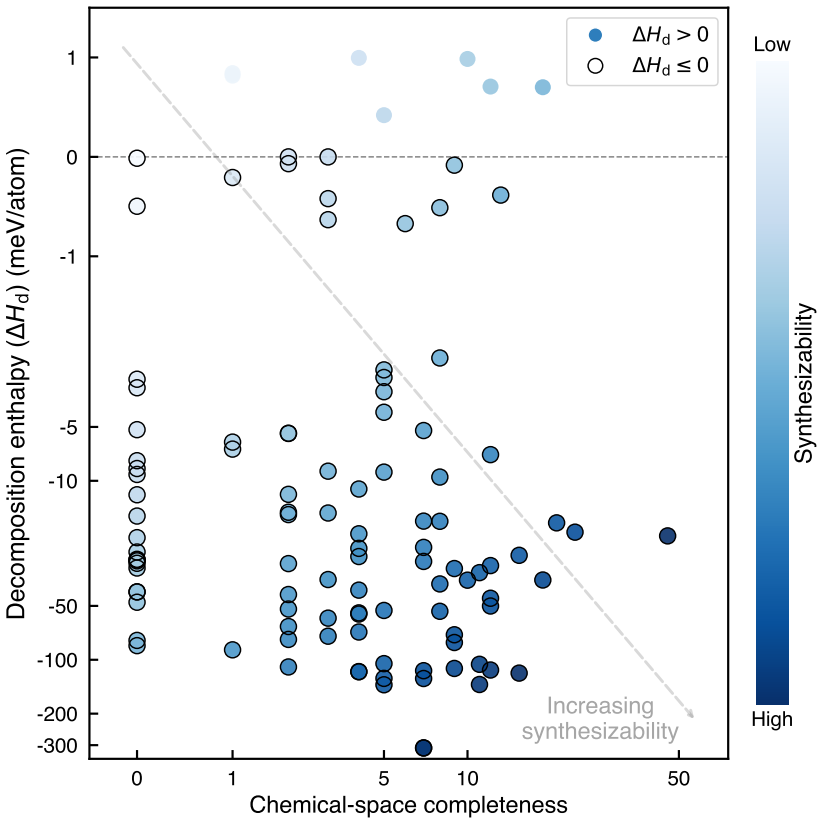
- Prioritization by decomposition enthalpy and chemical-space completeness identifies exactly 25 high-confidence synthesis targets.
Where Pith is reading between the lines
- The same three-stage protocol could be applied to other large databases of machine-learning-predicted materials to generate new experimental target lists.
- If the 25 candidates are successfully synthesized, it would support wider use of universal machine-learning potentials for dynamical pre-screening.
- Repeated failure to synthesize members of the final list would point to the need for more accurate or material-specific dynamical models.
Load-bearing premise
The universal machine-learning interatomic potentials used for dynamical screening are sufficiently accurate to identify materials that will remain stable under real experimental conditions.
What would settle it
Laboratory synthesis attempts on any of the 25 prioritized candidates that produce either a stable compound matching the predicted structure or clear decomposition would directly test whether the filtered list contains realizable materials.
Figures
read the original abstract
Machine-learning-accelerated materials discovery has yielded large numbers of computationally stable compounds, yet many remain experimentally unrealized, underscoring a persistent gap between prediction and synthesis. Here, we introduce a hierarchical screening framework that combines PBE-based thermodynamic stability, efficient dynamical-stability screening enabled by universal machine-learning interatomic potentials, and SCAN-based thermodynamic refinement. Applying this protocol to the 894 stable materials previously reported in Sci. Data 9, 302 (2022), we first curate 603 unique structures, of which only 298 remain thermodynamically stable on the complete PBE phase diagrams, demonstrating the critical role of competing phases in stability assessment. Dynamical screening then identifies 166 materials stable under both harmonic-phonon and finite-temperature molecular dynamics criteria, and SCAN phase diagrams further narrow this set to 109. Finally, by combining decomposition enthalpy with chemical-space completeness, we prioritize 25 candidates as high-confidence targets for experimental synthesis. This work provides a practical protocol for translating stability predictions into experimentally actionable synthesis targets, closing a key gap in machine-learning-driven materials discovery.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript presents a hierarchical screening protocol that applies PBE-based thermodynamic stability assessment (including full phase diagrams with competing phases), dynamical stability screening via universal machine-learning interatomic potentials (harmonic phonons plus finite-temperature MD), and SCAN-based thermodynamic refinement to an initial set of 894 materials from Sci. Data 9, 302 (2022). The protocol reduces the set to 603 unique structures, then 298 PBE-stable, 166 dynamically stable, 109 after SCAN, and finally prioritizes 25 high-confidence targets for experimental synthesis based on decomposition enthalpy and chemical-space completeness.
Significance. If the results hold, the work supplies a concrete, multi-stage filtering pipeline that translates large-scale computational stability predictions into a short list of synthesis targets, explicitly demonstrating the effect of competing phases and dynamical checks. The use of independent external benchmarks (PBE phase diagrams, ML potentials, SCAN) rather than internal circular definitions is a methodological strength, and the reporting of concrete reduction numbers at each stage aids reproducibility.
major comments (2)
- [abstract, dynamical screening paragraph] Abstract, dynamical screening paragraph: the reduction from 298 to 166 materials rests on harmonic-phonon and finite-T MD criteria computed exclusively with universal ML interatomic potentials, yet the manuscript provides no per-candidate DFT phonon benchmarks, convergence tests, or error estimates on imaginary-mode frequencies. Because this step is load-bearing for the subsequent 109 and final 25 candidates, the absence of validation directly limits in the high-confidence claim.
- [abstract, final prioritization sentence] Abstract, final prioritization sentence: the selection of the 25 candidates combines decomposition enthalpy with 'chemical-space completeness,' but the manuscript does not specify the quantitative definition or weighting of the completeness metric, nor does it show how it interacts with the SCAN decomposition enthalpies to produce the ranked list.
minor comments (1)
- [abstract] The abstract states that 603 unique structures were curated from the original 894; a brief description of the deduplication criteria (space-group tolerance, composition matching, etc.) would improve clarity.
Simulated Author's Rebuttal
We thank the referee for the constructive comments and positive assessment of the work's significance. We address each major comment below and will revise the manuscript accordingly to improve clarity and rigor.
read point-by-point responses
-
Referee: [abstract, dynamical screening paragraph] Abstract, dynamical screening paragraph: the reduction from 298 to 166 materials rests on harmonic-phonon and finite-T MD criteria computed exclusively with universal ML interatomic potentials, yet the manuscript provides no per-candidate DFT phonon benchmarks, convergence tests, or error estimates on imaginary-mode frequencies. Because this step is load-bearing for the subsequent 109 and final 25 candidates, the absence of validation directly limits in the high-confidence claim.
Authors: We agree that the dynamical screening step is critical and that the current manuscript lacks explicit per-candidate DFT validation. In the revised manuscript we will add a dedicated validation subsection (and corresponding supplementary tables) reporting DFT phonon calculations on a representative subset of ~25 candidates drawn from the 166. These will include convergence tests, direct comparison of imaginary-mode frequencies, and quantitative error estimates between the ML potentials and DFT. This addition will directly support the reliability of the 166 and downstream selections without changing the overall protocol or conclusions. revision: partial
-
Referee: [abstract, final prioritization sentence] Abstract, final prioritization sentence: the selection of the 25 candidates combines decomposition enthalpy with 'chemical-space completeness,' but the manuscript does not specify the quantitative definition or weighting of the completeness metric, nor does it show how it interacts with the SCAN decomposition enthalpies to produce the ranked list.
Authors: We thank the referee for highlighting this omission. The manuscript will be revised to provide an explicit quantitative definition of the chemical-space completeness metric (a normalized diversity score based on elemental composition vectors relative to the training database), the weighting scheme used to combine it with SCAN decomposition enthalpy (a linear combination with equal weights after normalization), and a supplementary table that shows the ranking procedure and the 25 selected candidates. The abstract will also be updated to reference this definition. These changes will make the prioritization fully reproducible. revision: yes
Circularity Check
No circularity; external dataset filtered by independent standard methods
full rationale
The derivation begins with the externally published set of 894 materials (Sci. Data 9, 302, 2022) and applies successive independent filters: PBE phase-diagram stability, ML-potential dynamical screening, and SCAN refinement. These steps use established external computational protocols rather than any quantity defined or fitted inside the present paper's equations. No self-citation is load-bearing for the central claim, no parameter is fitted to a subset and then relabeled a prediction, and no ansatz or uniqueness result is smuggled via prior author work. The final list of 25 is therefore a selection, not a reduction by construction.
Axiom & Free-Parameter Ledger
axioms (2)
- domain assumption PBE and SCAN functionals provide reliable relative thermodynamic stabilities when full phase diagrams are constructed.
- domain assumption Universal machine-learning interatomic potentials can accurately screen dynamical stability via harmonic phonons and finite-temperature MD.
Reference graph
Works this paper leans on
-
[1]
Nature materials , volume=
Computational high-throughput screening of electrocatalytic materials for hydrogen evolution , author=. Nature materials , volume=. 2006 , publisher=
2006
-
[2]
Chemistry of Materials , volume=
Phosphates as lithium-ion battery cathodes: an evaluation based on high-throughput ab initio calculations , author=. Chemistry of Materials , volume=. 2011 , publisher=
2011
-
[3]
Nature materials , volume=
The high-throughput highway to computational materials design , author=. Nature materials , volume=. 2013 , publisher=
2013
-
[4]
Nature Computational Science , volume=
A universal graph deep learning interatomic potential for the periodic table , author=. Nature Computational Science , volume=. 2022 , publisher=
2022
-
[5]
Nature , volume=
Scaling deep learning for materials discovery , author=. Nature , volume=. 2023 , publisher=
2023
-
[6]
npj Computational Materials , volume=
Predicting aqueous stability of solid with computed Pourbaix diagram using SCAN functional , author=. npj Computational Materials , volume=. 2020 , publisher=
2020
-
[7]
Chemistry of Materials , volume=
Artificial intelligence driving materials discovery? perspective on the article: Scaling deep learning for materials discovery , author=. Chemistry of Materials , volume=. 2024 , publisher=
2024
-
[8]
Journal of Materials Science , volume=
Review of computational approaches to predict the thermodynamic stability of inorganic solids , author=. Journal of Materials Science , volume=. 2022 , publisher=
2022
-
[9]
npj Computational Materials , volume=
Efficient first-principles prediction of solid stability: Towards chemical accuracy , author=. npj Computational Materials , volume=. 2018 , publisher=
2018
-
[10]
Advances in Neural Information Processing Systems , volume=
MACE: Higher order equivariant message passing neural networks for fast and accurate force fields , author=. Advances in Neural Information Processing Systems , volume=
-
[11]
npj Computational Materials , volume=
Universal machine learning interatomic potentials are ready for phonons , author=. npj Computational Materials , volume=. 2025 , publisher=
2025
-
[12]
Physical review B , volume=
Projector augmented-wave method , author=. Physical review B , volume=. 1994 , publisher=
1994
-
[13]
Physical Review B—Condensed Matter and Materials Physics , volume=
Oxidation energies of transition metal oxides within the GGA+ U framework , author=. Physical Review B—Condensed Matter and Materials Physics , volume=. 2006 , publisher=
2006
-
[14]
Physical Review B—Condensed Matter and Materials Physics , volume=
Formation enthalpies by mixing GGA and GGA+ U calculations , author=. Physical Review B—Condensed Matter and Materials Physics , volume=. 2011 , publisher=
2011
-
[15]
Computer Physics Communications , volume=
PHON: A program to calculate phonons using the small displacement method , author=. Computer Physics Communications , volume=. 2009 , publisher=
2009
-
[16]
APL materials , volume=
Commentary: The Materials Project: A materials genome approach to accelerating materials innovation , author=. APL materials , volume=. 2013 , publisher=
2013
-
[17]
2023 , publisher=
Understanding molecular simulation: from algorithms to applications , author=. 2023 , publisher=
2023
-
[18]
Chemical Science , volume=
Autonomous intelligent agents for accelerated materials discovery , author=. Chemical Science , volume=. 2020 , publisher=
2020
-
[19]
Scientific Data , volume=
Novel inorganic crystal structures predicted using autonomous simulation agents , author=. Scientific Data , volume=. 2022 , publisher=
2022
-
[20]
Physical review B , volume=
Ab initio molecular dynamics for liquid metals , author=. Physical review B , volume=. 1993 , publisher=
1993
-
[21]
Computational materials science , volume=
Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set , author=. Computational materials science , volume=. 1996 , publisher=
1996
-
[22]
Computational Materials Science , volume=
Python Materials Genomics (pymatgen): A robust, open-source python library for materials analysis , author=. Computational Materials Science , volume=. 2013 , publisher=
2013
-
[23]
Inorganic Crystal Structure Database
Karlsruhe, FIZ. Inorganic Crystal Structure Database
-
[24]
Physical review letters , volume=
Generalized gradient approximation made simple , author=. Physical review letters , volume=. 1996 , publisher=
1996
-
[25]
Physical review letters , volume=
Strongly constrained and appropriately normed semilocal density functional , author=. Physical review letters , volume=. 2015 , publisher=
2015
-
[26]
Journal of Physics: Condensed Matter , volume=
The atomic simulation environment—a Python library for working with atoms , author=. Journal of Physics: Condensed Matter , volume=. 2017 , publisher=
2017
-
[27]
Selective review of offline change point detection methods , journal =
Charles Truong and Laurent Oudre and Nicolas Vayatis , keywords =. Selective review of offline change point detection methods , journal =. 2020 , issn =. doi:https://doi.org/10.1016/j.sigpro.2019.107299 , url =
-
[28]
Acta Crystallographica Section A: Foundations of Crystallography , volume=
Crystal fingerprint space--a novel paradigm for studying crystal-structure sets , author=. Acta Crystallographica Section A: Foundations of Crystallography , volume=. 2010 , publisher=
2010
-
[29]
Proceedings of the National Academy of Sciences , volume=
Melting temperature prediction using a graph neural network model: From ancient minerals to new materials , author=. Proceedings of the National Academy of Sciences , volume=. 2022 , publisher=
2022
-
[30]
Physical Review Materials , volume=
Quantifying uncertainty in high-throughput density functional theory: A comparison of AFLOW, Materials Project, and OQMD , author=. Physical Review Materials , volume=. 2023 , publisher=
2023
-
[31]
npj Computational Materials , volume=
The role of decomposition reactions in assessing first-principles predictions of solid stability , author=. npj Computational Materials , volume=. 2019 , publisher=
2019
-
[32]
arXiv preprint arXiv:2504.06231 , year=
Orb-v3: atomistic simulation at scale , author=. arXiv preprint arXiv:2504.06231 , year=
-
[33]
Chemistry of Materials , volume=
Li- Fe- P- O2 phase diagram from first principles calculations , author=. Chemistry of Materials , volume=. 2008 , publisher=
2008
-
[34]
AI-Driven Expansion and Application of the Alexandria Database
AI-Driven Expansion and Application of the Alexandria Database , author=. arXiv preprint arXiv:2512.09169 , year=
work page internal anchor Pith review Pith/arXiv arXiv
-
[35]
Nature , volume=
A generative model for inorganic materials design , author=. Nature , volume=. 2025 , publisher=
2025
-
[36]
Joule , volume=
Mining unexplored chemistries for phosphors for high-color-quality white-light-emitting diodes , author=. Joule , volume=. 2018 , publisher=
2018
-
[37]
Nature , volume=
Machine learning for molecular and materials science , author=. Nature , volume=. 2018 , publisher=
2018
-
[38]
Juelsholt, Mikkel. Continued challenges in high-throughput materials predictions: MatterGen predicts compounds from the training dataset. Mater. Horiz. 2026. doi:10.1039/D6MH00268D
-
[39]
Materials Today Energy , pages=
Accelerating complex materials discovery with universal machine-learning potential-driven structure prediction , author=. Materials Today Energy , pages=. 2025 , publisher=
2025
-
[40]
ACS applied materials & interfaces , volume=
Energy, phonon, and dynamic stability criteria of two-dimensional materials , author=. ACS applied materials & interfaces , volume=. 2019 , publisher=
2019
-
[41]
Inorganic Chemistry , volume=
Search for Stable and Low-Energy Ce--Co--Cu Ternary Compounds Using Machine Learning , author=. Inorganic Chemistry , volume=. 2025 , publisher=
2025
-
[42]
Pseudo-potentials , author =
-
[43]
2002 , publisher=
Handbook on the physics and chemistry of rare earths , author=. 2002 , publisher=
2002
-
[44]
ACS Applied Energy Materials , volume=
Exploring Ultralow Lattice Thermal Conductivity in a 2D Hexagonal Structure of M3Te2 (M= Zn, Cd, and Hg) , author=. ACS Applied Energy Materials , volume=. 2025 , publisher=
2025
-
[45]
2000 , publisher=
Phase diagrams for binary alloys , author=. 2000 , publisher=
2000
-
[46]
Physical review letters , volume=
Crystal structure and pair potentials: A molecular-dynamics study , author=. Physical review letters , volume=. 1980 , publisher=
1980
-
[47]
arXiv preprint arXiv:2606.16385 , year=
Melt-Quench Failures and Practical Solutions for Universal Machine-Learning Interatomic Potentials in Amorphous Structure Generation , author=. arXiv preprint arXiv:2606.16385 , year=
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.