Streaming Molecular Dynamics Simulation Data for On-the-fly Processing and Analysis
Pith reviewed 2026-06-30 14:08 UTC · model grok-4.3
The pith
A new streaming framework gives molecular dynamics simulations direct real-time access to every generated time step instead of writing subsampled trajectories to disk.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
By extending the Interactive Molecular Dynamics protocol to IMDv3 and implementing it across GROMACS, NAMD, and LAMMPS, the framework streams every atomic position, velocity, and force generated during a running simulation directly to user-defined analysis routines, eliminating the need to discard most frames due to storage limits.
What carries the argument
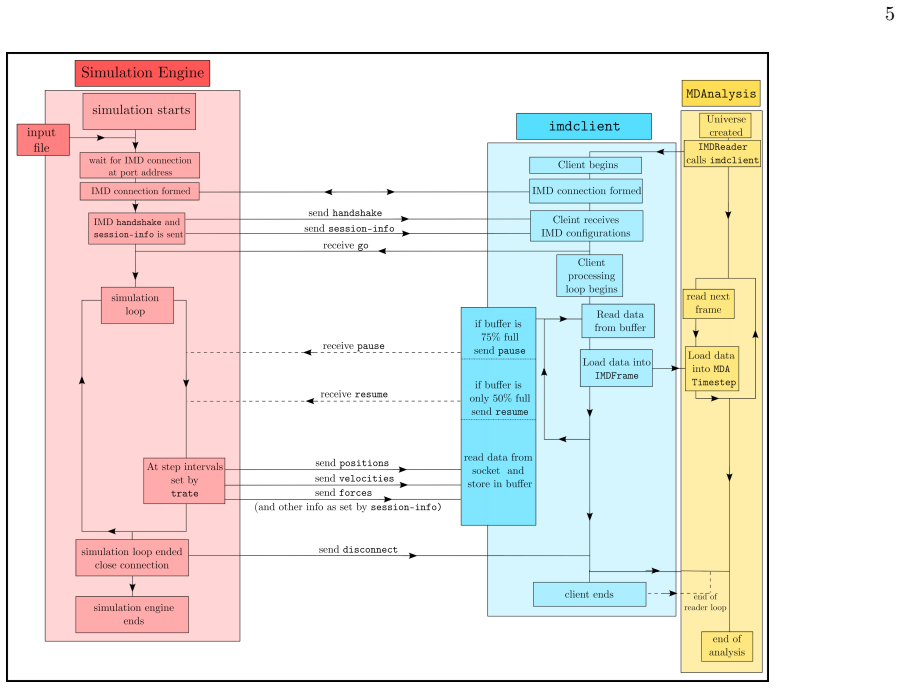
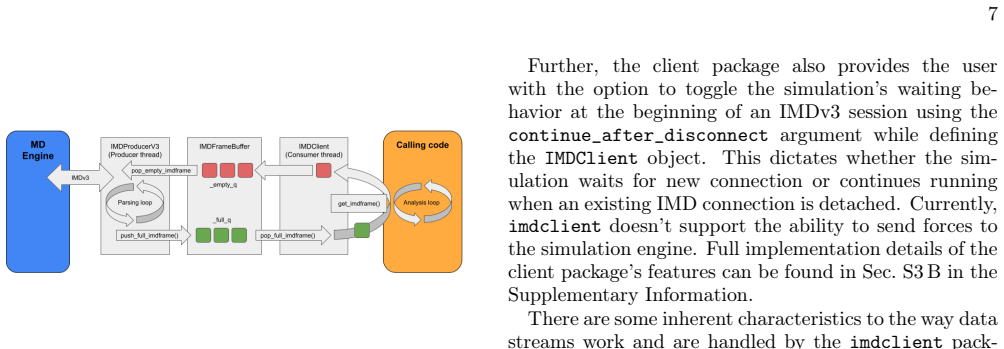
The IMDv3 protocol extension, which carries full per-step simulation data over a network stream to the imdclient Python package for immediate processing and MDAnalysis integration.
If this is right
- All femtosecond-scale data becomes available for analysis of molecular vibrations, solvent dynamics, and short-lived transition states.
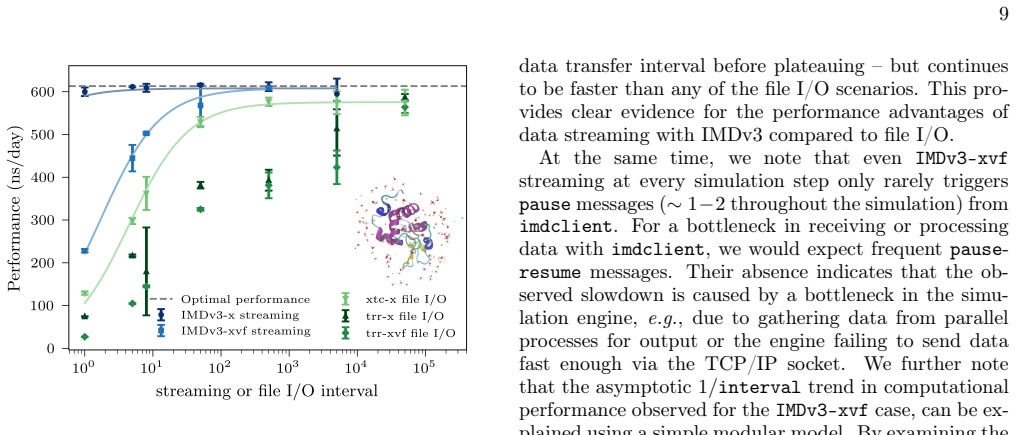
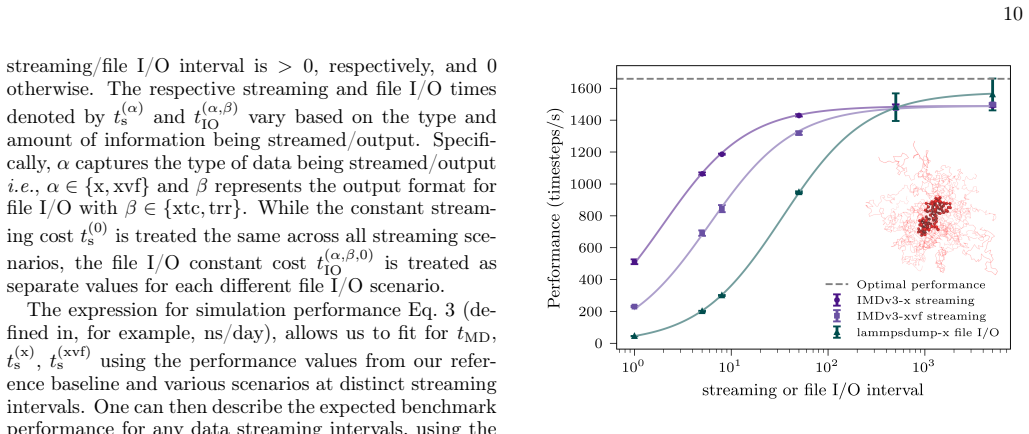
- Performance improves relative to simulations that write high-frequency trajectories to disk.
- Live monitoring of user-defined variables and real-time evaluation of velocity time-correlation functions become possible during the run.
- Real-time analysis of membrane pore currents and other transport phenomena can occur without post-processing large files.
Where Pith is reading between the lines
- The same streaming layer could support adaptive simulations that adjust parameters based on live analysis results.
- Storage requirements for long simulations drop because full trajectories need never be written.
- Coupling to external machine-learning tools becomes straightforward for on-the-fly property prediction.
- The approach may extend naturally to other MD engines once the IMDv3 interface is adopted.
Load-bearing premise
The extended IMDv3 protocol can be implemented reliably inside GROMACS, NAMD, and LAMMPS so that every generated data point reaches external analysis code in real time without data loss or prohibitive overhead.
What would settle it
A side-by-side run of the same multi-microsecond simulation once with IMDv3 streaming and once with femtosecond-interval trajectory writes; if the streamed run drops frames or shows higher wall-clock time than the disk-writing run, the performance and completeness claims are falsified.
Figures


read the original abstract
Only a small fraction of the data generated in state-of-the-art all-atom multi-microsecond molecular dynamics (MD) simulations is typically analyzed. With femtosecond integration steps, microsecond simulations generate billions of time steps containing atomic positions, velocities, and forces, often corresponding to petabytes of data. Since this exceeds practical storage capacities, only a subset of frames is usually written to trajectory files at intervals of 10-100 ps. While sufficient for ensemble averages and slow dynamics, this approach discards information on fast processes such as molecular vibrations, solvent dynamics, short-lived transition states, and transport phenomena that are directly related to macroscopic properties and experimental observables. Here, we introduce a streaming framework for MD simulations that provides direct access to all data generated during a running simulation. Instead of writing high-frequency output to disk, the framework enables user-defined analysis and processing routines to access simulation data in real time. To achieve this, we extend the Interactive Molecular Dynamics (IMD) protocol and implement an enhanced version, termed IMDv3, in the MD packages GROMACS, NAMD, and LAMMPS. We further introduce the Python package imdclient, which receives IMDv3 data streams and exposes them to external applications. To maximize usability, we add an imdclient-based reader to the MDAnalysis package, enabling streamed simulation data to be analyzed alongside conventional trajectory files. Benchmark results show that streaming can improve performance compared to simulations with high-frequency trajectory output. Example applications include live monitoring of custom variables, evaluation of velocity time-correlation functions with fast fluctuations, and real-time analysis of membrane pore currents.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The paper introduces a streaming framework for all-atom MD simulations that extends the IMD protocol to IMDv3, with implementations in GROMACS, NAMD, and LAMMPS. It provides the imdclient Python package to receive and expose the full data stream (positions, velocities, forces at every timestep) to external analysis code, plus an MDAnalysis reader for seamless integration with existing workflows. The central claim is that this enables real-time, lossless access to all generated data—avoiding subsampled disk trajectories—while improving performance over high-frequency output and supporting analysis of fast processes such as vibrations and transport.
Significance. If the implementations prove reliable and lossless at production rates, the approach would allow routine analysis of femtosecond-scale phenomena currently discarded, reduce storage demands, and shift MD workflows toward on-the-fly processing. The MDAnalysis integration lowers the barrier for adoption.
major comments (3)
- [Abstract and §3] Abstract and §3 (Implementation): the statement that 'implementations exist and benchmarks show performance gains' is not accompanied by any quantitative timing data, system sizes, timestep rates, or error bars on data completeness. Without these, the performance and 'all data reaches imdclient' claims cannot be evaluated.
- [§4] §4 (Protocol extension): the description of new IMDv3 message types does not specify buffering strategy, flow control, or handling of network latency at femtosecond output rates for systems of 10^4–10^5 atoms. This leaves the core assumption of zero data loss unverified.
- [§5] §5 (Benchmarks and examples): the velocity time-correlation and pore-current examples are presented without comparison to the same analysis performed on a subsampled trajectory or with reported frame-drop rates, making it impossible to confirm that the streaming advantage is realized in practice.
minor comments (2)
- [§2] Notation for timestep indexing and message sequence numbers should be defined explicitly in a table or equation to avoid ambiguity when discussing completeness.
- [Figure 2] Figure captions for benchmark plots should include the exact hardware, network configuration, and MD engine versions used.
Simulated Author's Rebuttal
We thank the referee for the constructive comments, which identify areas where additional quantitative details and clarifications would strengthen the manuscript. We address each major comment below and will incorporate revisions to provide the requested information and verifications.
read point-by-point responses
-
Referee: [Abstract and §3] Abstract and §3 (Implementation): the statement that 'implementations exist and benchmarks show performance gains' is not accompanied by any quantitative timing data, system sizes, timestep rates, or error bars on data completeness. Without these, the performance and 'all data reaches imdclient' claims cannot be evaluated.
Authors: We agree that the abstract and §3 currently lack the specific quantitative benchmarks needed to evaluate the performance claims. In the revised manuscript we will add detailed timing results, including system sizes (e.g., 10^4–10^5 atoms), timestep rates, wall-clock comparisons with high-frequency disk output, and measured data completeness (frame delivery rates with error bars) from the GROMACS, NAMD, and LAMMPS implementations. revision: yes
-
Referee: [§4] §4 (Protocol extension): the description of new IMDv3 message types does not specify buffering strategy, flow control, or handling of network latency at femtosecond output rates for systems of 10^4–10^5 atoms. This leaves the core assumption of zero data loss unverified.
Authors: The referee is correct that §4 describes the new message types but does not detail the buffering, flow-control, or latency-handling mechanisms. We will expand §4 with a description of the client-side and server-side buffering strategy, TCP flow control, and any rate-limiting or back-pressure mechanisms employed to maintain lossless delivery at femtosecond output rates for the cited system sizes. revision: yes
-
Referee: [§5] §5 (Benchmarks and examples): the velocity time-correlation and pore-current examples are presented without comparison to the same analysis performed on a subsampled trajectory or with reported frame-drop rates, making it impossible to confirm that the streaming advantage is realized in practice.
Authors: We acknowledge that the examples in §5 illustrate the workflow but omit direct quantitative comparisons to subsampled trajectories and explicit frame-drop statistics. In the revision we will add these comparisons (where the same analysis can be performed on both streamed and subsampled data) together with measured frame-drop rates to demonstrate that the streaming approach preserves the fast-process information that would otherwise be lost. revision: yes
Circularity Check
No circularity: software framework with no derivations or self-referential predictions
full rationale
The paper describes implementation of an extended IMDv3 protocol in GROMACS/NAMD/LAMMPS plus the imdclient package and MDAnalysis integration. It contains no mathematical derivations, fitted parameters, uniqueness theorems, or predictions that reduce to inputs by construction. Performance claims rest on empirical benchmarks rather than any self-definitional or fitted-input structure. No load-bearing self-citations or ansatzes are present. The central contribution is a practical engineering extension whose validity is externally testable via code and runtime measurements.
Axiom & Free-Parameter Ledger
Reference graph
Works this paper leans on
-
[1]
topol.tpr
IMDv3-enabled MDAnalysis The most evident use-case for IMDv3 streamed data is data analysis. To facilitate this application in a seam- less manner, we implemented a corresponding Reader class within MDAnalysis [38], a popular Python package to read and analyze simulation data. The IMDReader class enables the user to useMDAnalysis’s API to load and process...
-
[2]
Running simulations with these two versions, varying the number of cores used, over a com- bination of thread-MPI tasks and OpenMP threads, tells us two important things
GROMACS We consider versions of GROMACS pre and post- IMDv3 modifications, which we call the ‘vanilla’ and ‘imdv3’ versions. Running simulations with these two versions, varying the number of cores used, over a com- bination of thread-MPI tasks and OpenMP threads, tells us two important things. Evidently, under the same con- ditions the IMDv3-modified ver...
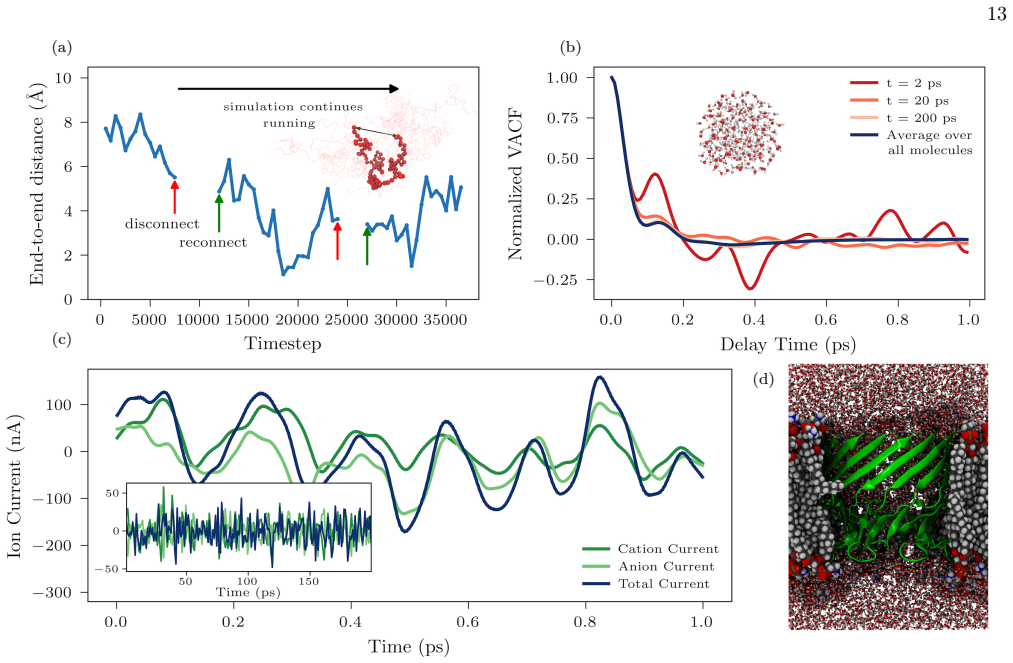
-
[3]
For NAMD, we use the same biomolec- ular system as described forGROMACS
LAMMPS & NAMD We perform similar benchmarks for LAMMPS and NAMD - comparing performance when using streaming with file I/O. For NAMD, we use the same biomolec- ular system as described forGROMACS. However, for LAMMPS, we choose a system that more closely resem- bles typical use cases in the corresponding community. Here, we use a simple FENE-bonded polyme...
-
[4]
E. R. Lindahl,Molecular modeling of proteins (Springer,
-
[5]
S. A. Hollingsworth and R. O. Dror, Molecular Dynamics Simulation for All, Neuron99, 1129 (2018)
2018
-
[6]
Frenkel and B
D. Frenkel and B. Smit,Understanding molecular simu- lation: from algorithms to applications (Elsevier, 2023)
2023
-
[7]
Warshel, Molecular dynamics simulations of biological reactions, Accounts of Chemical Research35, 385 (2002)
A. Warshel, Molecular dynamics simulations of biological reactions, Accounts of Chemical Research35, 385 (2002)
2002
-
[8]
S.A.AdcockandJ.A.McCammon,Moleculardynamics: survey of methods for simulating the activity of proteins, Chemical Reviews106, 1589 (2006)
2006
-
[9]
D. J. Huggins, P. C. Biggin, M. A. Dämgen, J. W. Essex, S. A. Harris, R. H. Henchman, S. Khalid, A. Kuzmanic, C. A. Laughton, J. Michel,et al., Biomolecular simula- tions: From dynamics and mechanisms to computational assays of biological activity, Wiley Interdisciplinary Re- views: ComputationalMolecularScience 9,e1393(2019)
2019
-
[10]
Axelrod, D
S. Axelrod, D. Schwalbe-Koda, S. Mohapatra, J. Dame- wood, K. P. Greenman, and R. Gómez-Bombarelli, Learning matter: Materials design with machine learn- ing and atomistic simulations, Accounts of Materials Re- search 3, 343 (2022)
2022
-
[11]
Maginn and J
E. Maginn and J. Elliott, Historical perspective and cur- rent outlook for molecular dynamics as a chemical en- gineering tool, Industrial & Engineering Chemistry Re- search 49, 3059 (2010)
2010
-
[12]
Karmakar, A
T. Karmakar, A. R. Finney, M. Salvalaglio, A. O. Yazaydin, and C. Perego, Non-equilibrium modeling of concentration-driven processes with constant chemical potential molecular dynamics simulations, Accounts of Chemical Research56, 1156 (2023)
2023
-
[13]
Henzler-Wildman and D
K. Henzler-Wildman and D. Kern, Dynamic personalities of proteins, Nature450, 964 (2007)
2007
-
[14]
G. R. Bowman, Accurately modeling nanosecond protein dynamics requires at least microseconds of simulation, Journal of Computational Chemistry37, 558 (2016)
2016
-
[15]
D. E. Shaw, P. J. Adams, A. Azaria, J. A. Bank, B. Bat- son, A. Bell, M. Bergdorf, J. Bhatt, J. A. Butts, T. Cor- reia, et al., Anton 3: Twenty microseconds of molecular dynamics simulation before lunch, inProceedings of the International Conference for High Performance Comput- ing, Networking, Storage and Analysis (2021) pp. 1–11
2021
-
[16]
Heyden, Heterogeneity of water structure and dynam- ics at the protein-water interface, Journal of Chemical Physics 150, 094701 (2019)
M. Heyden, Heterogeneity of water structure and dynam- ics at the protein-water interface, Journal of Chemical Physics 150, 094701 (2019)
2019
-
[17]
Foster, M
I. Foster, M. Ainsworth, B. Allen, J. Bessac, F. Cap- pello, J. Y. Choi, E. Constantinescu, P. E. Davis, S. Di, W. Di, H. Guo, S. Klasky, K. K. Van Dam, T. Kurc, Q. Liu, A. Malik, K. Mehta, K. Mueller, T. Munson, G. Ostouchov, M. Parashar, T. Peterka, L. Pouchard, D. Tao, O. Tugluk, S. Wild, M. Wolf, J. M. Wozniak, W. Xu, and S. Yoo, Computing Just What Y...
2017
-
[18]
Beckstein, G
O. Beckstein, G. Fox, and S. Jha, Convergence of data generation and analysis in the biomolecular simula- tion community, in Online Resource for Big Data and Extreme-Scale Computing Workshop (2018) p. 4
2018
-
[19]
Heyden and M
M. Heyden and M. Havenith, Combining thz spec- troscopy and md simulations to study protein-hydration coupling, Methods52, 74 (2010)
2010
-
[20]
Heyden, J
M. Heyden, J. Sun, S. Funkner, G. Mathias, H. Forbert, M. Havenith, and D. Marx, Dissecting the THz spectrum of liquid water from first principles via correlations in time and space, Proceedings of the National Academy of Sciences 107, 12068 (2010)
2010
-
[21]
M. A. Sauer and M. Heyden, Frequency-Selective Anhar- monic Mode Analysis of Thermally Excited Vibrations in Proteins, Journal of Chemical Theory and Computation 19, 5481 (2023)
2023
-
[22]
R. A. X. Persson, V. Pattni, A. Singh, S. M. Kast, and M. Heyden, Signatures of solvation thermodynamics in spectra of intermolecular vibrations, Journal of Chemical Theory and Computation13, 4467 (2017)
2017
-
[23]
Beckstein and M
O. Beckstein and M. S. P. Sansom, Liquid–vapor oscilla- tions of water in hydrophobic nanopores, Proceedings of the National Academy of Sciences100, 7063 (2003)
2003
-
[24]
Beckstein and M
O. Beckstein and M. S. P. Sansom, The influence of ge- ometry, surface character, and flexibility on the perme- ation of ions and water through biological pores, Physical Biology 1, 42 (2004)
2004
-
[25]
J. D. Russo, S. Zhang, J. M. G. Leung, A. T. Bogetti, J. P. Thompson, A. J. DeGrave, P. A. Torrillo, A. J. Pratt, K. F. Wong, J. Xia, J. Copperman, J. L. Adelman, M. C. Zwier, D. N. LeBard, D. M. Zuckerman, and L. T. Chong, WESTPA 2.0: High-Performance Upgrades for Weighted Ensemble Simulations and Analysis of Longer- Timescale Applications, Journal of Ch...
2022
-
[26]
S. D. Schwartz and V. L. Schramm, Enzymatic transition states and dynamic motion in barrier crossing, Nature Chemical Biology5, 551 (2009)
2009
-
[27]
J. C. Bennett, H. Abbasi, P.-T. Bremer, R. Grout, A. Gyulassy, T. Jin, S. Klasky, H. Kolla, M. Parashar, V. Pascucci, P. Pebay, D. Thompson, H. Yu, F. Zhang, and J. Chen, Combining In-Situ and in-Transit Process- ing to Enable Extreme-Scale Scientific Analysis, inPro- ceedings of the International Conference on High Per- formance Computing, Networking, St...
2012
-
[28]
Lakshminarasimhan, D
S. Lakshminarasimhan, D. A. Boyuka, S. V. Pendse, X. Zou, J. Jenkins, V. Vishwanath, M. E. Papka, and N. F. Samatova, Scalable in situ scientific data encod- ing for analytical query processing, inProceedings of the 22nd international symposium on High-performance par- allel and distributed computing , HPDC ’13 (Association for Computing Machinery, New Yo...
2013
-
[29]
J. E. Stone, J. Gullingsrud, and K. Schulten, A system for interactive molecular dynamics simulation, in Pro- ceedings of the 2001 symposium on Interactive 3D graph- ics, I3D ’01 (Association for Computing Machinery, New York, NY, USA, 2001) pp. 191–194
2001
-
[30]
Kampfrath, R
M. Kampfrath, R. Staritzbichler, G. P. Hernández, A. S. Rose, J. K. S. Tiemann, G. Scheuermann, D. Wiegreffe, and P. W. Hildebrand, MDsrv: visual sharing and analy- sis of molecular dynamics simulations, Nucleic Acids Re- search 50, W483 (2022)
2022
-
[31]
Dreher and B
M. Dreher and B. Raffin, A Flexible Framework for 15 Asynchronous in Situ and in Transit Analytics for Sci- entific Simulations, in 2014 14th IEEE/ACM Interna- tional Symposium on Cluster, Cloud and Grid Comput- ing (Chicago, IL, USA, 2014) pp. 277–286
2014
-
[32]
Johnston, B
T. Johnston, B. Zhang, A. Liwo, S. Crivelli, and M. Taufer, In situ data analytics and indexing of pro- tein trajectories, Journal of Computational Chemistry 38, 1419 (2017)
2017
-
[33]
Malakar, C
P. Malakar, C. Knight, T. Munson, V. Vishwanath, and M. E. Papka, Scalable In situ Analysis of Molecular Dy- namicsSimulations,in ISA V’17 Proceedings of the In Situ Infrastructures on Enabling Extreme-Scale Analysis and Visualization (Denver, CO, USA, 2017) pp. 1–6
2017
-
[34]
H. C. Zanúz, B. Raffin, O. A. Mures, and E. J. Padrón, In-transitmoleculardynamicsanalysiswithApacheflink, in Proceedings of the Workshop on In Situ Infrastructures for Enabling Extreme-Scale Analysis and Visualization (ACM, Dallas Texas USA, 2018) pp. 25–32
2018
-
[35]
Grayson, J
P. Grayson, J. Gullingsrud, K. Schulten, and J. E. Stone, Interactive Molecular Dynamics Simulation (2013)
2013
-
[36]
Lanrezac, N
A. Lanrezac, N. Férey, and M. Baaden, Interactive Molecular Dynamics, in Comprehensive Computational Chemistry (Elsevier, 2024) pp. 454–474
2024
-
[37]
S. Páll, A. Zhmurov, P. Bauer, M. Abraham, M. Lund- borg, A. Gray, B. Hess, and E. Lindahl, Heterogeneous parallelization and acceleration of molecular dynamics simulations in GROMACS, Journal of Chemical Physics 153, 134110 (2020)
2020
-
[38]
J. C. Phillips, D. J. Hardy, J. D. C. Maia, J. E. Stone, J. V. Ribeiro, R. C. Bernardi, R. Buch, G. Fiorin, J. Hénin, W. Jiang, R. McGreevy, M. C. R. Melo, B. K. Radak, R. D. Skeel, A. Singharoy, Y. Wang, B. Roux, A. Aksimentiev, Z. Luthey-Schulten, L. V. Kalé, K. Schulten, C. Chipot, and E. Tajkhorshid, Scal- able molecular dynamics on CPU and GPU archit...
2020
-
[39]
A. P. Thompson, H. M. Aktulga, R. Berger, D. S. Bolin- tineanu, W. M. Brown, P. S. Crozier, P. J. in ’t Veld, A. Kohlmeyer, S. G. Moore, T. D. Nguyen, R. Shan, M. J. Stevens, J. Tranchida, C. Trott, and S. J. Plimpton, LAMMPS - a flexible simulation tool for particle-based materials modeling at the atomic, meso, and continuum scales, Computer Physics Comm...
2022
-
[40]
Humphrey, A
W. Humphrey, A. Dalke, and K. Schulten, VMD – Vi- sual Molecular Dynamics, J Molecular Modelling14, 33 (1996)
1996
-
[41]
R. J. Gowers, M. Linke, J. Barnoud, T. J. E. Reddy, M. N. Melo, S. L. Seyler, D. L. Dotson, J. Domanski, S. Buchoux, I. M. Kenney, and O. Beckstein, MDAnaly- sis: A Python package for the rapid analysis of molecular dynamics simulations., inProceedings of the 15th Python in Science Conference, edited by S. Benthall and S. Ros- trup (Austin, TX, 2016) pp. 102–109
2016
-
[42]
L. J. Woods, O. Beckstein, A. Thirumalaiswamy, and M. Heyden, IMDv3 Protocol (2025)
2025
-
[43]
Woods, A
L. Woods, A. Thirumalaiswamy, H. Cho, H. MacDermott-Opeskin, J. A. Clark, M. Heyden, and O. Beckstein, imdclient (2025)
2025
-
[44]
Gowers, O
R. Gowers, O. Beckstein, M. Linke, I. Alibay, J. Barnoud, T. Reddy, H. MacDermott-Opeskin, R. Meli, L. Wang, U. Bansal, J. Zeman, J. Detlefs, D. L. Dotson, M. N. Melo, M. Bieniek, ayushsuhane, S. Buchoux, M. Tib- erti, N. Michaud-Agrawal, D. Cruz, S. Seyler, denniej0 2, P. Loche, J. Borreguero, Y. Zhuang, T. Yelgoe, N. Pal, Ninad, J. Domański, and Z. Wu, ...
2025
-
[45]
Woods, A
L. Woods, A. Thirumalaiswamy, H. Cho, O. Beckstein, and M. Heyden, modified version ofGROMACS 2024.4 with imdv3 support (2026)
2024
-
[46]
Johansson, E
A. Johansson, E. Weinberg, C. Trott, M. McCarthy, and S. Moore, Lammps-kokkos: Performance portable molec- ular dynamics across exascale architectures, inProceed- ings of the SC ’25 Workshops of the International Con- ference for High Performance Computing, Networking, Storage and Analysis , SC Workshops ’25 (Association for Computing Machinery, New York,...
2025
-
[47]
Woods, A
L. Woods, A. Thirumalaiswamy, O. Beckstein, and M. Heyden, modified version ofLAMMPS with support for data streaming via imdv3 (2026)
2026
-
[48]
J. C. Phillips, D. J. Hardy, J. D. C. Maia, J. E. Stone, J. V. Ribeiro, R. C. Bernardi, R. Buch, G. Fiorin, J. Hénin, W. Jiang, R. McGreevy, M. C. R. Melo, B. K. Radak, R. D. Skeel, A. Singharoy, Y. Wang, B. Roux, A. Aksimentiev, Z. Luthey-Schulten, L. V. Kalé, K. Schulten, C. Chipot, and E. Tajkhorshid, Scal- able molecular dynamics on cpu and gpu archit...
2020
-
[49]
Woods, H
L. Woods, H. MacDermott-Opeskin, E. Jakupovic, Y. Zhuang, R. J. Gowers, and O. Beckstein, Zarrtraj: A Pythonpackageforstreamingmoleculardynamicstrajec- tories from cloud services, Journal of Open Source Soft- ware 11, 7943 (2026)
2026
-
[50]
D. M. Jennewein, J. Lee, C. Kurtz, W. Dizon, I. Sha- effer, A. Chapman, A. Chiquete, J. Burks, A. Carlson, N. Mason, A. Kobawala, T. Jagadeesan, P. B. Basani, T. Battelle, R. Belshe, D. McCaffrey, M. Brazil, C. Inu- mella, K. Kuznia, J. Buzinski, D. D. Shah, S. M. Dud- ley, G. Speyer, and J. Yalim, The sol supercomputer at arizona state university, in Pra...
2023
-
[51]
Thirumalaiswamy, Heydenlabasu-collab/imdv3- performance-tests: Benchmarking of molecular dynamics simulations with streaming vs
A. Thirumalaiswamy, Heydenlabasu-collab/imdv3- performance-tests: Benchmarking of molecular dynamics simulations with streaming vs. file i/o (2026)
2026
-
[52]
Thirumalaiswamy, Heydenlabasu-collab/imdv3- applications: Example applications of molecular dynamics data streaming with imdv3 (2026)
A. Thirumalaiswamy, Heydenlabasu-collab/imdv3- applications: Example applications of molecular dynamics data streaming with imdv3 (2026)
2026
-
[53]
Eddy, Transmission control protocol (tcp) (2022)
W. Eddy, Transmission control protocol (tcp) (2022)
2022
-
[54]
Taylor,An Introduction to Error Analysis: The Study of Uncertainties in Physical Measurements (MIT Press, 2022)
J. Taylor,An Introduction to Error Analysis: The Study of Uncertainties in Physical Measurements (MIT Press, 2022). S1 Supporting Information: Streaming Molecular Dynamics Simulation Data for On-the-fly Processing and Analysis S1. TERMS Below is a list of terms and their definitions as used in the main and SI text. IMD: Interactive Molecular Dynamics – a ...
2022
-
[55]
out of order
Packet Types In IMD-specific data communication, data is sent in the form of message packets, with each message typically containing two sub-packets viz. a header packet and a body packet. A header packet is composed of8 bytes. The first 4 bytes contain the header type and the next 4 bytes serve as a flexible slot for holding other information. The header...
-
[56]
Blocking: Wait until a receiver is connected to begin execution of the simulation
-
[57]
Non-blocking: Begin the simulation regardless of whether a receiver is connected and continuously check on the listening socket for a receiver attempting to connect The simulation engine’s waiting behavior also applies when a receiver disconnects mid-simulation:
-
[58]
Blocking: Pause simulation execution and wait until a receiver is connected to resume execution
-
[59]
Non-blocking: Continue execution, continuously checking on the listening socket for a receiver attempting to connect Header: 16 (int32) Wait <val> (int32) Nonzero to set the simulation engine’s waiting behavior to blocking, 0 to set the simulation engine’s waiting behavior to non-blocking Note: The purpose of this packet is to allow a receiver to monitor ...
-
[60]
The data within each IMD frame is always sent in the same, fixed order:
Packet order After the simulation engine sends thehandshake and session info packets to the receiver and gets back ago signal, it begins sending simulation data via IMD. The data within each IMD frame is always sent in the same, fixed order:
-
[61]
Note: In IMDv3 implementations, the simulation engine and client require that all packets specified in session info must be sent for every IMD frame and in the same order
Forces If the simulation engine is configured to send only a strict subset of all available data packets, the fixed order of the list still applies to the remaining packets in the session. Note: In IMDv3 implementations, the simulation engine and client require that all packets specified in session info must be sent for every IMD frame and in the same ord...
-
[62]
GROMACS To run IMDv3 inGROMACSone has to make the following changes to the molecular dynamics parameter (*.mdp) input file: 1 ; Required IMD settings 2 IMD-group = System 3 ; Atom group to stream (System = all atoms) 4 5 IMD-nst = 100 S8 6 ; Send data every 100 steps 7 IMD-version = 3 8 ; Use IMDv3 protocol (2/3) - defaults to 2 9 10 ; Data types and spec...
-
[63]
LAMMPS In LAMMPS, one adds the following line to the input file to enable IMDv3 functionality: 1 # IMD configuration 2 3 # fix ID group-ID imd <port> version <2/3> nowait <on/off> trate <arg> time <yes/no> box <yes/no> ,→ coordinates <yes/no> velocities <yes/no> forces <yes/no> unwrap <yes/no> 4 fix imdv3 all imd 8888 nowait off trate 100 version 3 time y...
-
[64]
>" for big and
NAMD Finally, forNAMD one can similarly edit the input file with the following settings: 1 # Required IMD settings 2 IMDon yes ; Enable IMD functionality (yes/no) 3 IMDport 8888 ; Port number for socket connection 4 IMDwait on ; Wait for client connection before starting simulation (on/off) 5 IMDfreq 100 ; Send data every 100 steps (transmission rate) 6 I...
-
[65]
6a), polymer ID-number1 (randomly chosen for the example) is monitored by calculating its end-to-end distance with appropriate periodic boundary conditions (PBC) based corrections
End-to-end distance of polymer In this example (see Fig. 6a), polymer ID-number1 (randomly chosen for the example) is monitored by calculating its end-to-end distance with appropriate periodic boundary conditions (PBC) based corrections. The exact calculation can be found in theIMDv3-applications repository. lend-end = ∥r1 − rN ∥pbc (S1) where r1 and rN a...
-
[66]
V ACF of water Here (see Fig. 6b), the average velocity autocorrelation function (VACF) of a single water molecule in the system is calculated over a fixed lag time period and visualized at various simulation times. VACF here is defined as, VACFi(τ) = ⟨vi(t) · vi(t + τ)⟩t ⟨vi(t) · vi(t)⟩t (S2) where vi(t) is the velocity of theith water molecule at timet,...
-
[67]
Ion Current through membrane Here (see Fig. 6c), we calculate and visualize the ion current (nA) across the membrane pore, defined as follows: Ii(t) = 1 Lz∆t NiX j=1 qj (zj(t + ∆t) − zj(t))pbc (S3) where Ii(t) is the ion current from a certain ion group like cations, anions or all ions. These ion currents are calculated across a time interval∆t in the z-d...
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.