Chemical Origins of Non-Bonded Interactions Within and Between Solids
Pith reviewed 2026-05-19 14:56 UTC · model grok-4.3
pith:4C7TFEXM Add to your LaTeX paper
What is a Pith Number?\usepackage{pith}
\pithnumber{4C7TFEXM}
Prints a linked pith:4C7TFEXM badge after your title and writes the identifier into PDF metadata. Compiles on arXiv with no extra files. Learn more
The pith
A generalization of ALMO-EDA to solids decomposes non-bonded interactions into frozen, polarization, and charge transfer contributions.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
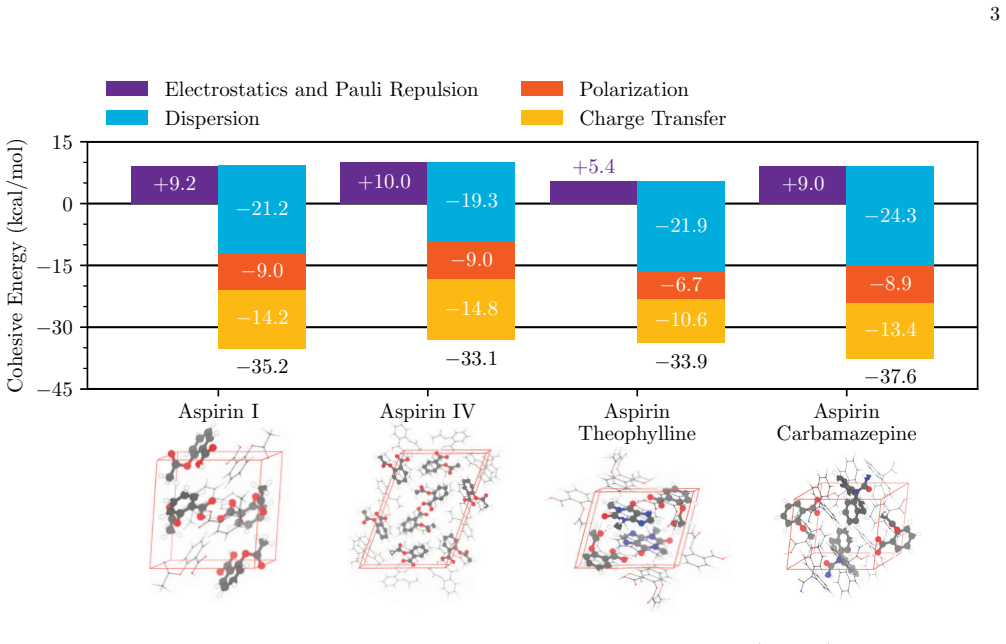
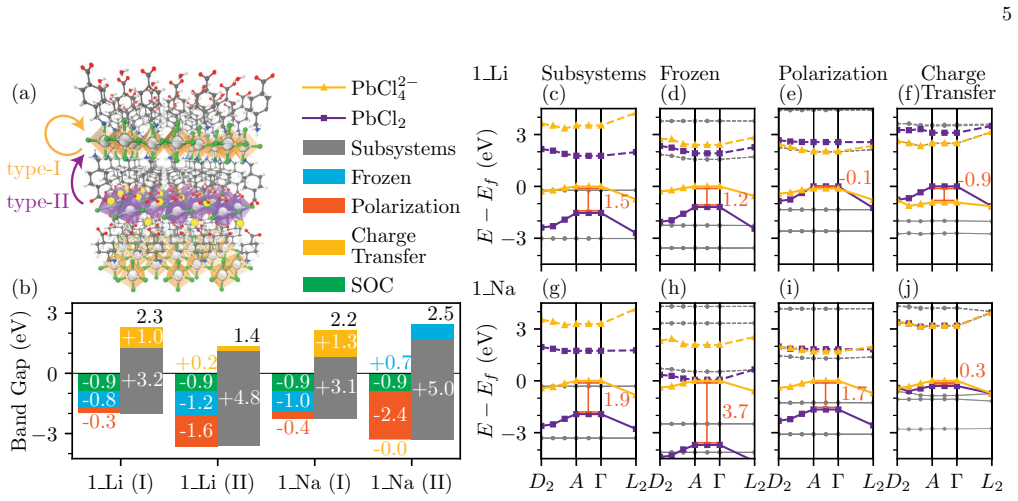
The central discovery is the periodic extension of ALMO-EDA that quantifies non-bonded interactions within and between solids by decomposing them into frozen interactions, polarization, and charge transfer at the DFT level. This is demonstrated across molecular crystals where dispersion controls polymorph stability, in MoS2/WSe2 where stacking modulates interlayer coupling, and in layered perovskite heterostructures where alkali cation substitution switches quantum-well character.
What carries the argument
The generalized ALMO-EDA method for periodic boundary conditions, which partitions the interaction energies and band structure changes into frozen, polarization, and charge-transfer terms to maintain chemical interpretability.
Load-bearing premise
That the generalization to periodic boundary conditions preserves the chemical interpretability of the frozen, polarization, and charge transfer terms without significant artifacts for the systems studied.
What would settle it
Observing that for a known test case like two interacting molecules in a periodic cell, the decomposed energy terms do not match the expected chemical contributions from gas-phase calculations or total energy differences.
Figures


read the original abstract
Non-bonded interactions govern structure, stability, and function across a wide range of solid-state materials, yet their chemical origins are often difficult to resolve from total energies alone. Here we generalize absolutely localized molecular orbital energy decomposition analysis to quantify and interpret non-bonded interactions within and between solids at the density functional theory level. Across molecular crystals, moir\'e heterobilayers, and layered perovskite heterostructures, this framework separates lattice-formation energies, interlayer binding energies, and band-structure changes into chemically intuitive contributions from frozen interactions, polarization, and charge transfer. The analysis reveals how dispersion controls polymorph stability in pharmaceutical crystals, how local stacking modulates interlayer coupling in MoS2/WSe2, and how alkali-cation substitution switches the quantum-well character of layered perovskite heterostructures. By connecting emergent solid-state properties to microscopic interaction mechanisms, this framework provides a chemically transparent basis for understanding and designing complex materials.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript generalizes absolutely localized molecular orbital energy decomposition analysis (ALMO-EDA) to periodic boundary conditions at the DFT level. It applies the framework to molecular crystals, moiré heterobilayers (e.g., MoS2/WSe2), and layered perovskite heterostructures, decomposing lattice-formation energies, interlayer binding energies, and band-structure changes into frozen interactions, polarization, and charge transfer. The analysis links these terms to properties such as dispersion-controlled polymorph stability, stacking-modulated interlayer coupling, and alkali-cation effects on quantum-well character.
Significance. If the periodic extension preserves clean separation of the EDA terms without PBC-induced mixing of charge transfer with band dispersion, the work supplies a chemically transparent route to interpret non-bonded interactions in extended solids. This could aid rational design in pharmaceuticals, 2D heterostructures, and perovskites by connecting microscopic contributions to emergent behaviors.
major comments (1)
- [Methods / periodic ALMO-EDA implementation] Periodic generalization of ALMO-EDA: the central claim requires that the decomposition cleanly isolates frozen, polarization, and charge-transfer contributions. In PBC the absolutely localized orbitals become Wannier-like or fragment-projected Bloch states, and the charge-transfer term arises by relaxing the localization constraint. This risks entanglement with intrinsic band dispersion and interlayer hybridization already present in the isolated fragments, especially in the MoS2/WSe2 moiré and alkali-substituted perovskite cases where band-structure changes are reported. An explicit validation against independent charge-density differences or electrostatic-potential maps is needed to confirm the separation retains its chemical meaning.
minor comments (2)
- [Results / applications] A table summarizing the decomposed energy components (frozen, polarization, CT) for each material class would improve quantitative comparison across the reported systems.
- [Computational details] The manuscript should state the specific DFT functional, dispersion correction, and k-point sampling used in all calculations to support reproducibility.
Simulated Author's Rebuttal
We thank the referee for their careful reading of the manuscript and for the constructive comment on the periodic ALMO-EDA implementation. We address the concern regarding clean separation of terms below and have incorporated the suggested validation.
read point-by-point responses
-
Referee: Periodic generalization of ALMO-EDA: the central claim requires that the decomposition cleanly isolates frozen, polarization, and charge-transfer contributions. In PBC the absolutely localized orbitals become Wannier-like or fragment-projected Bloch states, and the charge-transfer term arises by relaxing the localization constraint. This risks entanglement with intrinsic band dispersion and interlayer hybridization already present in the isolated fragments, especially in the MoS2/WSe2 moiré and alkali-substituted perovskite cases where band-structure changes are reported. An explicit validation against independent charge-density differences or electrostatic-potential maps is needed to confirm the separation retains its chemical meaning.
Authors: We appreciate the referee's emphasis on rigorously confirming the separation of EDA terms under periodic boundary conditions. In the periodic ALMO-EDA implementation, each fragment (individual molecules for crystals or single layers for heterostructures) is represented by its own set of fragment-projected Bloch states obtained from a self-consistent calculation on the isolated fragment within the periodic cell. The frozen term is evaluated from the antisymmetrized product of these fragment densities interacting through the full periodic potential, thereby incorporating any intrinsic band dispersion of the isolated fragments. Polarization allows intra-fragment orbital relaxation within each fragment's subspace in response to the other fragments. The charge-transfer contribution is isolated as the further energy lowering upon removal of the inter-fragment localization constraint. This hierarchical construction ensures that band dispersion and hybridization intrinsic to the isolated fragments remain in the reference or polarization steps rather than contaminating the CT term. For the MoS2/WSe2 moiré and alkali-substituted perovskite examples, band-structure changes are decomposed by comparing eigenvalues of the full system against those of the polarized fragments, with the CT term specifically capturing additional interlayer orbital mixing. To directly address the request for validation, we have added explicit comparisons in the revised manuscript between the CT densities and independent charge-density difference maps together with electrostatic potential shifts for these two systems; the maps confirm that the CT term corresponds to the expected interlayer charge redistribution without conflation with dispersion or intra-fragment hybridization. revision: yes
Circularity Check
No significant circularity detected in the ALMO-EDA generalization to periodic solids
full rationale
The paper describes a methodological extension of absolutely localized molecular orbital energy decomposition analysis (ALMO-EDA) to periodic boundary conditions at the DFT level. The central framework separates lattice-formation energies, interlayer binding energies, and band-structure changes into frozen interactions, polarization, and charge transfer terms using the standard definitions and relaxation steps of the EDA approach. No equations, self-citations, or fitted parameters are presented that reduce any reported separation or prediction to an input by construction. The applications to molecular crystals, MoS2/WSe2 heterobilayers, and layered perovskites are analyses performed with the generalized method rather than tautological outputs. The derivation chain remains self-contained against external benchmarks of the original ALMO-EDA formalism and does not rely on load-bearing self-references or ansatzes smuggled through citations.
Axiom & Free-Parameter Ledger
axioms (1)
- domain assumption Density functional theory calculations accurately capture the non-bonded interactions in the studied solid systems.
Lean theorems connected to this paper
-
IndisputableMonolith/Cost/FunctionalEquation.leanwashburn_uniqueness_aczel unclear?
unclearRelation between the paper passage and the cited Recognition theorem.
We generalize absolutely localized molecular orbital energy decomposition analysis (ALMO-EDA) to periodic systems... E_int^AB ≡ E_frz + E_pol + E_ct
-
IndisputableMonolith/Foundation/BranchSelection.leanbranch_selection unclear?
unclearRelation between the paper passage and the cited Recognition theorem.
The frozen term contains classical electrostatics, Pauli repulsion, and dispersion... polarization... charge-transfer
What do these tags mean?
- matches
- The paper's claim is directly supported by a theorem in the formal canon.
- supports
- The theorem supports part of the paper's argument, but the paper may add assumptions or extra steps.
- extends
- The paper goes beyond the formal theorem; the theorem is a base layer rather than the whole result.
- uses
- The paper appears to rely on the theorem as machinery.
- contradicts
- The paper's claim conflicts with a theorem or certificate in the canon.
- unclear
- Pith found a possible connection, but the passage is too broad, indirect, or ambiguous to say the theorem truly supports the claim.
Reference graph
Works this paper leans on
-
[1]
Stone, Anthony , month = jan, year =. The
-
[2]
WIREs Computational Molecular Science , author =
Recent developments in symmetry‐adapted perturbation theory , volume =. WIREs Computational Molecular Science , author =. 2020 , pages =. doi:10.1002/wcms.1452 , abstract =
-
[3]
Mo, Yirong and Bao, Peng and Gao, Jiali , year =. Energy decomposition analysis based on a block-localized wavefunction and multistate density functional theory , volume =. Physical Chemistry Chemical Physics , publisher =. doi:10.1039/C0CP02206C , abstract =
-
[4]
The Journal of Chemical Physics , author =
Perspective:. The Journal of Chemical Physics , author =. 2017 , pages =. doi:10.1063/1.4978951 , abstract =
-
[5]
and Das, Akshaya and Demerdash, Omar and Levine, Daniel S
Mao, Yuezhi and Loipersberger, Matthias and Horn, Paul R. and Das, Akshaya and Demerdash, Omar and Levine, Daniel S. and Prasad Veccham, Srimukh and Head-Gordon, Teresa and Head-Gordon, Martin , year =. From. Annual Review of Physical Chemistry , publisher =. doi:https://doi.org/10.1146/annurev-physchem-090419-115149 , abstract =
-
[6]
Morokuma, Keiji , month = aug, year =. Molecular. The Journal of Chemical Physics , publisher =. doi:10.1063/1.1676210 , abstract =
-
[7]
A new energy decomposition scheme for molecular interactions within the
Kitaura, Kazuo and Morokuma, Keiji , month = mar, year =. A new energy decomposition scheme for molecular interactions within the. International Journal of Quantum Chemistry , publisher =. doi:10.1002/qua.560100211 , abstract =
-
[8]
Mo, Yirong and Gao, Jiali and Peyerimhoff, Sigrid D. , month = apr, year =. Energy decomposition analysis of intermolecular interactions using a block-localized wave function approach , volume =. The Journal of Chemical Physics , publisher =. doi:10.1063/1.481185 , abstract =
-
[9]
Theoretica chimica acta , author =
On the calculation of bonding energies by the. Theoretica chimica acta , author =. 1977 , pages =. doi:10.1007/BF02401406 , abstract =
-
[10]
Theoretica Chimica Acta , author =
On the use of local basis sets for localized molecular orbitals , volume =. Theoretica Chimica Acta , author =. 1980 , pages =. doi:10.1007/BF00574903 , language =
-
[11]
International Journal of Quantum Chemistry , author =
Modification of the. International Journal of Quantum Chemistry , author =. 1996 , pages =. doi:10.1002/(SICI)1097-461X(1996)60:1<157::AID-QUA17>3.0.CO;2-C , abstract =
-
[12]
Mo, Yirong and Peyerimhoff, Sigrid D. , month = aug, year =. Theoretical analysis of electronic delocalization , volume =. The Journal of Chemical Physics , publisher =. doi:10.1063/1.476742 , abstract =
-
[13]
Khaliullin, Rustam Z. and Cobar, Erika A. and Lochan, Rohini C. and Bell, Alexis T. and Head-Gordon, Martin , month = sep, year =. Unravelling the. The Journal of Physical Chemistry A , publisher =. doi:10.1021/jp073685z , abstract =
-
[14]
The Journal of Chemical Physics , author =
Unrestricted absolutely localized molecular orbitals for energy decomposition analysis:. The Journal of Chemical Physics , author =. 2013 , pages =. doi:10.1063/1.4798224 , abstract =
-
[15]
The Journal of Chemical Physics , author =
Polarization contributions to intermolecular interactions revisited with fragment electric-field response functions , volume =. The Journal of Chemical Physics , author =. 2015 , pages =. doi:10.1063/1.4930534 , abstract =
-
[16]
Philipsen, P. H. T. and Baerends, E. J. , month = jun, year =. Role of the. The Journal of Physical Chemistry B , publisher =. doi:10.1021/jp060886e , abstract =
-
[17]
The Journal of Chemical Physics , author =
A periodic energy decomposition analysis method for the investigation of chemical bonding in extended systems , volume =. The Journal of Chemical Physics , author =. 2015 , pages =. doi:10.1063/1.4919943 , abstract =
-
[18]
Staub, Ruben and Iannuzzi, Marcella and Khaliullin, Rustam Z. and Steinmann, Stephan N. , month = jan, year =. Energy. Journal of Chemical Theory and Computation , publisher =. doi:10.1021/acs.jctc.8b00957 , abstract =
-
[19]
Journal of the American Chemical Society , author =
Tuning the. Journal of the American Chemical Society , author =. 2025 , pages =. doi:10.1021/jacs.5c08391 , abstract =
-
[20]
Crystal. Chemical Reviews , author =. 2022 , pages =. doi:10.1021/acs.chemrev.1c00987 , abstract =
-
[21]
Advanced capabilities for materials modelling with
Giannozzi, P and Andreussi, O and Brumme, T and Bunau, O and Buongiorno Nardelli, M and Calandra, M and Car, R and Cavazzoni, C and Ceresoli, D and Cococcioni, M and Colonna, N and Carnimeo, I and Dal Corso, A and de Gironcoli, S and Delugas, P and DiStasio, R A and Ferretti, A and Floris, A and Fratesi, G and Fugallo, G and Gebauer, R and Gerstmann, U an...
-
[22]
The Journal of Chemical Physics , author =
Software for the frontiers of quantum chemistry:. The Journal of Chemical Physics , author =. 2021 , pages =. doi:10.1063/5.0055522 , abstract =
-
[23]
and Deems, Stephen and Furlani, Thomas R
Boerner, Timothy J. and Deems, Stephen and Furlani, Thomas R. and Knuth, Shelley L. and Towns, John , year =. Practice and. doi:10.1145/3569951.3597559 , abstract =
-
[24]
Perdew, John P. and Burke, Kieron and Ernzerhof, Matthias , month = oct, year =. Generalized. Phys. Rev. Lett. , publisher =. doi:10.1103/PhysRevLett.77.3865 , number =
-
[25]
The Journal of Chemical Physics , author =
Efficient all-electron periodic. The Journal of Chemical Physics , author =. 2026 , pages =. doi:10.1063/5.0303084 , abstract =
-
[26]
Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for
Weigend, Florian and Ahlrichs, Reinhart , year =. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for. Physical Chemistry Chemical Physics , publisher =. doi:10.1039/B508541A , abstract =
-
[27]
and Altarawy, Doaa and Didier, Brett and Gibson, Tara D
Pritchard, Benjamin P. and Altarawy, Doaa and Didier, Brett and Gibson, Tara D. and Windus, Theresa L. , month = nov, year =. New. Journal of Chemical Information and Modeling , publisher =. doi:10.1021/acs.jcim.9b00725 , number =
-
[28]
Chemical Physics Letters , author =. 1998 , pages =. doi:10.1016/S0009-2614(98)00862-8 , abstract =
-
[29]
A geometric approach to direct minimization , volume =
Van Voorhis, Troy and Head-Gordon, Martin , month = jun, year =. A geometric approach to direct minimization , volume =. Molecular Physics , publisher =. doi:10.1080/00268970110103642 , abstract =
-
[30]
Boys, S.F. and Bernardi, F. , month = oct, year =. The calculation of small molecular interactions by the differences of separate total energies. Molecular Physics , publisher =. doi:10.1080/00268977000101561 , number =
-
[31]
The Journal of Chemical Physics , author =
Influence of the exchange screening parameter on the performance of screened hybrid functionals , volume =. The Journal of Chemical Physics , author =. 2006 , pages =. doi:10.1063/1.2404663 , abstract =
-
[32]
The Journal of Chemical Physics , author =
Generalized gradient approximation model exchange holes for range-separated hybrids , volume =. The Journal of Chemical Physics , author =. 2008 , pages =. doi:10.1063/1.2921797 , abstract =
-
[33]
The Journal of Chemical Physics , author =
Gaussian basis sets for accurate calculations on molecular systems in gas and condensed phases , volume =. The Journal of Chemical Physics , author =. 2007 , pages =. doi:10.1063/1.2770708 , abstract =
-
[34]
Cobar, Erika A. and Horn, Paul R. and Bergman, Robert G. and Head-Gordon, Martin , year =. Examination of the hydrogen-bonding networks in small water clusters (n = 2–5, 13, 17) using absolutely localized molecular orbital energy decomposition analysis , volume =. Physical Chemistry Chemical Physics , publisher =. doi:10.1039/C2CP42522J , abstract =
-
[35]
and Mao, Yuezhi and Head-Gordon, Martin , year =
Horn, Paul R. and Mao, Yuezhi and Head-Gordon, Martin , year =. Probing non-covalent interactions with a second generation energy decomposition analysis using absolutely localized molecular orbitals , volume =. Physical Chemistry Chemical Physics , publisher =. doi:10.1039/C6CP03784D , abstract =
-
[36]
Yang, Jun and Hu, Weifeng and Usvyat, Denis and Matthews, Devin and Schütz, Martin and Chan, Garnet Kin-Lic , month = aug, year =. Ab initio determination of the crystalline benzene lattice energy to sub-kilojoule/mole accuracy , volume =. Science , publisher =. doi:10.1126/science.1254419 , abstract =
-
[37]
The Journal of Physical Chemistry C , author =. 2014 , pages =. doi:10.1021/jp501237c , abstract =
-
[38]
Physical Chemistry Chemical Physics , author =
Practical quantum mechanics-based fragment methods for predicting molecular crystal properties , volume =. Physical Chemistry Chemical Physics , author =. 2012 , pages =. doi:10.1039/c2cp23949c , language =
-
[39]
Nature Communications , author =
Nucleic-acid-base photofunctional cocrystal for information security and antimicrobial applications , volume =. Nature Communications , author =. 2024 , pages =. doi:10.1038/s41467-024-46869-6 , abstract =
-
[40]
Co-crystal engineering of a two-dimensional perovskite phase for perovskite solar modules with improved efficiency and stability , volume =. Nature Energy , author =. 2025 , pages =. doi:10.1038/s41560-025-01903-9 , language =
-
[41]
Crystal Growth & Design , author =
A. Crystal Growth & Design , author =. 2018 , pages =. doi:10.1021/acs.cgd.8b01330 , abstract =
-
[42]
Nicolaï, Béatrice and Fournier, Bertrand and Dahaoui, Slimane and Gillet, Jean-Michel and Ghermani, Nour-Eddine , month = feb, year =. Crystal and. Crystal Growth & Design , publisher =. doi:10.1021/acs.cgd.8b01698 , number =
-
[43]
Shtukenberg, Alexander G. and Hu, Chunhua T. and Zhu, Qiang and Schmidt, Martin U. and Xu, Wenqian and Tan, Melissa and Kahr, Bart , month = jun, year =. The. Crystal Growth & Design , publisher =. doi:10.1021/acs.cgd.7b00673 , number =
-
[44]
Chemical & Pharmaceutical Bulletin , author =
Structure. Chemical & Pharmaceutical Bulletin , author =. 1985 , pages =. doi:10.1248/cpb.33.2641 , number =
-
[45]
A consistent and accurate ab initio parametrization of density functional dispersion correction (. The Journal of Chemical Physics , author =. 2010 , pages =. doi:10.1063/1.3382344 , abstract =
-
[46]
Effect of the damping function in dispersion corrected density functional theory , volume =
Grimme, Stefan and Ehrlich, Stephan and Goerigk, Lars , month = may, year =. Effect of the damping function in dispersion corrected density functional theory , volume =. Journal of Computational Chemistry , publisher =. doi:10.1002/jcc.21759 , abstract =
-
[47]
Novoselov, K. S. and Mishchenko, A. and Carvalho, A. and Castro Neto, A. H. , month = jul, year =. Science , publisher =. doi:10.1126/science.aac9439 , abstract =
-
[48]
Nature Nanotechnology , author =
Semiconductor moiré materials , volume =. Nature Nanotechnology , author =. 2022 , pages =. doi:10.1038/s41565-022-01165-6 , abstract =
-
[49]
Interlayer couplings,. Science Advances , author =. 2017 , pages =. doi:10.1126/sciadv.1601459 , abstract =
-
[50]
Chemical Communications , author =
Shape and size mimicry in the design of ternary molecular solids: towards a robust strategy for crystal engineering , volume =. Chemical Communications , author =. 2011 , pages =. doi:10.1039/c1cc14567c , language =
-
[51]
Desiraju, Gautam R. , month = jul, year =. Crystal. Journal of the American Chemical Society , publisher =. doi:10.1021/ja403264c , number =
-
[52]
Fleischman, Scott G. and Kuduva, Srinivasan S. and McMahon, Jennifer A. and Moulton, Brian and Bailey Walsh, Rosa D. and Rodríguez-Hornedo, Naír and Zaworotko, Michael J. , month = nov, year =. Crystal. Crystal Growth & Design , publisher =. doi:10.1021/cg034035x , number =
-
[53]
Directed assembly of layered perovskite heterostructures as single crystals , volume =. Nature , author =. 2021 , pages =. doi:10.1038/s41586-021-03810-x , language =
-
[54]
and Rettig, Adam and Dinh, Hieu Q
Robinson, Paul J. and Rettig, Adam and Dinh, Hieu Q. and Chen, Meng‐Fu and Lee, Joonho , month = jan, year =. Condensed‐. WIREs Computational Molecular Science , publisher =. doi:10.1002/wcms.70005 , abstract =
-
[55]
Nature Nanotechnology , author =
Excitons in semiconductor moiré superlattices , volume =. Nature Nanotechnology , author =. 2022 , pages =. doi:10.1038/s41565-021-01068-y , language =
-
[56]
The Journal of Chemical Physics , author =. 2020 , pages =. doi:10.1063/5.0007045 , abstract =
-
[57]
Symmetry-adapted perturbation theory of intermolecular forces , volume =
Szalewicz, Krzysztof , month = mar, year =. Symmetry-adapted perturbation theory of intermolecular forces , volume =. WIREs Computational Molecular Science , publisher =. doi:10.1002/wcms.86 , abstract =
-
[58]
Blöchl, P. E. , month = dec, year =. Projector augmented-wave method , volume =. Physical Review B , publisher =. doi:10.1103/PhysRevB.50.17953 , number =
-
[59]
Journal of Molecular Graphics , author =. 1996 , keywords =. doi:10.1016/0263-7855(96)00018-5 , abstract =
-
[60]
Nature Communications , author =
Electronic signature of the instantaneous asymmetry in the first coordination shell of liquid water , volume =. Nature Communications , author =. 2013 , pages =. doi:10.1038/ncomms2459 , abstract =
-
[61]
Niedzielski, Grzegorz and Hooper, James G. M. , month = sep, year =. A. Journal of Chemical Theory and Computation , publisher =. doi:10.1021/acs.jctc.5c01043 , number =
-
[62]
Imamura, Yutaka and Takahashi, Asuka and Okada, Takeshi and Ohno, Takahisa and Nakai, Hiromi , month = mar, year =. Extension of energy density analysis to periodic-boundary-condition calculations with plane-wave basis functions , volume =. Physical Review B , publisher =. doi:10.1103/PhysRevB.81.115136 , number =
-
[63]
Physical Review Letters , author =
Predicting. Physical Review Letters , author =. 2008 , pages =. doi:10.1103/PhysRevLett.101.115503 , language =
-
[64]
and Rettig, Adam and Lee, Joonho , month = nov, year =
Ni, Anton Z. and Rettig, Adam and Lee, Joonho , month = nov, year =. Gaussian-. Journal of Chemical Theory and Computation , publisher =. doi:10.1021/acs.jctc.5c01403 , number =
-
[65]
The Journal of Chemical Physics , author =
An energy decomposition analysis for second-order. The Journal of Chemical Physics , author =. 2015 , pages =. doi:10.1063/1.4929479 , abstract =
-
[66]
Filip, Marina R. and Qiu, Diana Y. and Del Ben, Mauro and Neaton, Jeffrey B. , month = jun, year =. Screening of. Nano Letters , publisher =. doi:10.1021/acs.nanolett.2c01306 , number =
-
[67]
Wang, Zhenling and Ikeda, Kevin and Shen, Hengyuan and Loipersberger, Matthias and Zech, Alexander and Aldossary, Abdulrahman and Head-Gordon, Teresa and Head-Gordon, Martin , month = feb, year =. Second-. Journal of Chemical Theory and Computation , publisher =. doi:10.1021/acs.jctc.4c01301 , number =
-
[68]
Perdew, John P. , month = mar, year =. Density functional theory and the band gap problem , volume =. International Journal of Quantum Chemistry , publisher =. doi:10.1002/qua.560280846 , abstract =
-
[69]
Computational Materials Science , author =
Band structure diagram paths based on crystallography , volume =. Computational Materials Science , author =. 2017 , keywords =. doi:10.1016/j.commatsci.2016.10.015 , abstract =
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.