Partially reactive force field for the UiO-66 metal-organic framework
Pith reviewed 2026-05-20 04:16 UTC · model grok-4.3
The pith
A new partially reactive force field models UiO-66 structure, stability, and early node-ligand assembly steps.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
nb-UiO-FF reproduces structural features of both UiO-66 and its isoreticular analog UiO-67, mechanical properties and framework stability with or without defects, activated or filled with N,N-dimethylformamide or ethanol. The force field is further employed within a molecular dynamics scheme to study the early stages of solvothermal node-ligand binding. Transient structural motifs both thermodynamically and kinetically favored are identified. This force field enables studying the self-assembly of UiO-66, as well as the formation of its point defects.
What carries the argument
The nb-UiO-FF force field, which adds a Morse potential to capture node-ligand reactivity and dummy atoms to represent the anisotropic charge distribution on zirconium atoms in the nodes.
If this is right
- The force field supports molecular dynamics studies of UiO-66 self-assembly.
- It identifies transient structural motifs that are both thermodynamically and kinetically favored during node-ligand binding.
- The same parameters correctly describe the isoreticular UiO-67 framework.
- Framework stability and mechanical properties remain accurate with or without defects and with different solvents.
- The model can now be used to investigate point defect formation during synthesis.
Where Pith is reading between the lines
- The same approach could be adapted to simulate synthesis of other zirconium MOFs that also form defects readily.
- The favored motifs point to possible experimental levers, such as solvent choice, for controlling defect levels.
- Longer simulations might show how these early arrangements grow into crystals or lead to disordered regions.
- The force field offers a route to predict how synthesis conditions affect final porosity and stability without full quantum calculations.
Load-bearing premise
The Morse potential parameters and dummy-atom charges can be chosen such that the resulting classical model accurately captures the node-ligand reactivity and anisotropic Zr charge distribution across the range of conditions studied.
What would settle it
Molecular dynamics runs with nb-UiO-FF that produce transient structural motifs different from those observed in spectroscopy or ab initio calculations of early node-ligand binding would show the model does not correctly describe assembly.
Figures







read the original abstract
UiO-66 is the most widely studied metal-organic framework (MOF), on account of its structural tunability given by its capacity of sustaining high amounts of point defects in its structure. Its synthesis mechanism is largely unknown, with only a few works mostly focused on the formation of the Zr-oxide cluster. In this work, a partially reactive force field to model UiO-66, nb-UiO-FF, is introduced. This force field incorporates node--ligand reactivity via a Morse potential and the introduction of dummy atoms to reproduce the anisotropic charge distribution of the Zr atoms in the node. nb-UiO-FF reproduces structural features of both UiO-66 and its isoreticular analog UiO-67, mechanical properties and framework stability with or without defects, activated or filled with N,N-dimethylformamide or ethanol. The force field is further employed within a molecular dynamics scheme to study the early stages of solvothermal node--ligand binding. Transient structural motifs both thermodynamically and kinetically favored are identified. This force field enables studying the self-assembly of UiO-66, as well as the formation of its point defects.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript introduces nb-UiO-FF, a partially reactive classical force field for UiO-66 that augments standard potentials with a Morse term for node-ligand reactivity and dummy atoms on Zr to capture anisotropic charge distribution. It reports that the model reproduces structural features of UiO-66 and the isoreticular UiO-67, mechanical properties, and framework stability both with and without defects and in the presence of DMF or ethanol. The force field is then used in molecular-dynamics simulations of solvothermal conditions to identify transient, thermodynamically and kinetically favored node-ligand binding motifs, with the broader goal of enabling studies of self-assembly and defect formation.
Significance. A transferable, reactive force field for UiO-66 would be a useful addition to the MOF modeling toolkit, particularly for exploring synthesis pathways and defect chemistry that are difficult to access with static DFT or non-reactive potentials. The combination of a Morse interaction with dummy-atom charges is a pragmatic way to introduce limited reactivity without moving to reactive bond-order potentials. If the parameterization proves robust outside the fitting set, the work could support falsifiable predictions about motif populations during early-stage assembly.
major comments (2)
- [Abstract / Parameterization] Abstract and parameter-fitting section: no quantitative validation metrics (RMSD for lattice parameters, percentage errors on bulk moduli, or defect formation energies) or error bars are supplied, nor is the target data set used to determine the Morse well depth, range, and dummy-atom charges described. Without these, it is impossible to judge whether the reported reproduction of structures, mechanics, and stability is within acceptable tolerances or merely qualitative.
- [MD Simulations of Node-Ligand Binding] MD results on transient motifs: the central claim that certain binding motifs are both thermodynamically and kinetically favored rests on the transferability of the fitted Morse parameters and dummy charges to non-equilibrium node-ligand approach trajectories. No independent test (e.g., comparison of approach energies or barriers to ab initio metadynamics or to a separate DFT data set) is reported, leaving open the possibility that the observed motif ranking is an artifact of the fitting procedure.
minor comments (2)
- [Methods] Notation for the dummy-atom charges and positions should be defined explicitly in a table or equation rather than only in the text.
- [Results] Figure captions for the motif snapshots should state the simulation temperature, solvent model, and time window over which the motif is observed.
Simulated Author's Rebuttal
We thank the referee for their constructive review and for recognizing the potential utility of a partially reactive force field for studying UiO-66 assembly and defects. We address each major comment below and have revised the manuscript accordingly where the points identify clear gaps in presentation or validation.
read point-by-point responses
-
Referee: [Abstract / Parameterization] Abstract and parameter-fitting section: no quantitative validation metrics (RMSD for lattice parameters, percentage errors on bulk moduli, or defect formation energies) or error bars are supplied, nor is the target data set used to determine the Morse well depth, range, and dummy-atom charges described. Without these, it is impossible to judge whether the reported reproduction of structures, mechanics, and stability is within acceptable tolerances or merely qualitative.
Authors: We agree that explicit quantitative metrics and a clear description of the fitting data are necessary for rigorous assessment. In the revised manuscript we have added a dedicated parameterization subsection that specifies the DFT-derived target data set (optimized node-ligand geometries, interaction energies, and partial charges) used to determine the Morse parameters and dummy-atom charges. We have also inserted a validation table reporting RMSD values for lattice parameters of UiO-66 and UiO-67, percentage deviations in bulk moduli and defect formation energies relative to reference DFT and experimental data, and standard deviations from replicate simulations. revision: yes
-
Referee: [MD Simulations of Node-Ligand Binding] MD results on transient motifs: the central claim that certain binding motifs are both thermodynamically and kinetically favored rests on the transferability of the fitted Morse parameters and dummy charges to non-equilibrium node-ligand approach trajectories. No independent test (e.g., comparison of approach energies or barriers to ab initio metadynamics or to a separate DFT data set) is reported, leaving open the possibility that the observed motif ranking is an artifact of the fitting procedure.
Authors: We acknowledge the referee’s concern about transferability to non-equilibrium trajectories. The force field was fitted exclusively to equilibrium structures and energies; its use in solvothermal MD therefore relies on the assumption that the Morse term remains physically reasonable during approach. While a full ab initio metadynamics benchmark was outside the scope of the present study, we have added to the revised manuscript (i) a limitations paragraph discussing this assumption and (ii) a supplementary comparison of binding energies extracted from selected MD snapshots against single-point DFT calculations on the same geometries. These additions provide partial independent support for the motif ranking while making the transferability caveat explicit. revision: partial
Circularity Check
No circularity: standard force-field parameterization with independent validation targets
full rationale
The paper introduces nb-UiO-FF by adding a Morse term for node-ligand reactivity and dummy atoms for Zr anisotropy, then reports that the resulting model reproduces equilibrium structures, moduli, defect stability, and solvent-filled behavior for UiO-66/67 while also generating new MD observations of transient binding motifs. No equation or claim in the abstract or described workflow equates a reported prediction to a fitted input by construction, nor does any load-bearing step rest on a self-citation chain that itself assumes the target result. The derivation therefore remains self-contained: parameters are chosen to match selected reference data, and the subsequent structural, mechanical, and dynamical results are presented as applications rather than tautological restatements of those choices.
Axiom & Free-Parameter Ledger
free parameters (2)
- Morse potential parameters for node-ligand interaction
- Dummy atom charges and positions on Zr
axioms (1)
- domain assumption Standard classical force-field terms (harmonic bonds, angles, Lennard-Jones non-bonded interactions) remain valid for all interactions not involving the added Morse term.
invented entities (1)
-
Dummy atoms attached to Zr nodes
no independent evidence
Reference graph
Works this paper leans on
-
[1]
Partially reactive force field for the UiO-66 metal-organic framework
and Quick-FF[27] were devised.[28] However, most force fields struggle to model the hydroxylated version of UiO-66, due to the complex interplay between intra- cluster and cluster-ligand interactions.[29] Most impor- tantly, these force fields are unable to capture reactive processes, since they operate over a pre-defined connec- tivity. This renders them...
work page internal anchor Pith review Pith/arXiv arXiv 2026
-
[2]
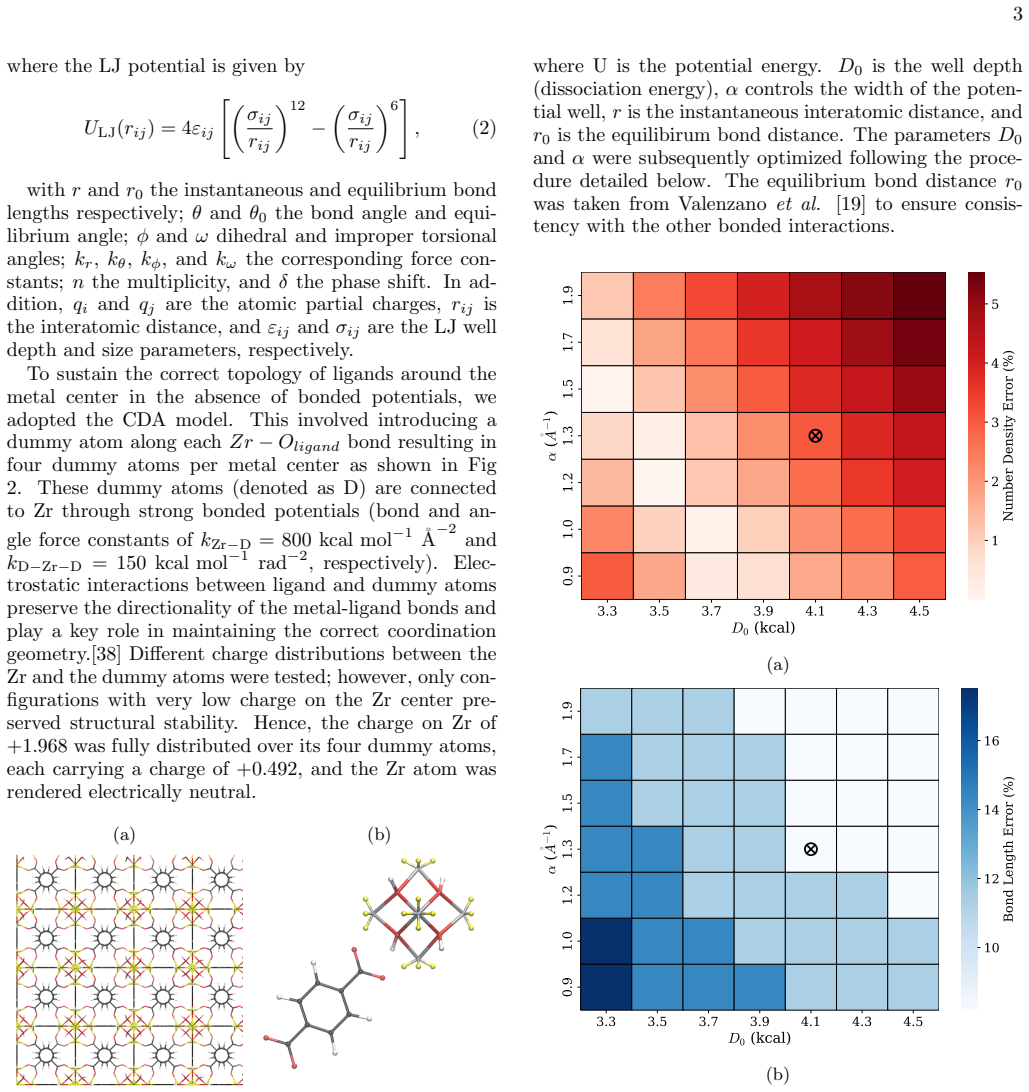
These dummy atoms (denoted as D) are connected to Zr through strong bonded potentials (bond and an- gle force constants of kZr−D = 800 kcal mol −1 ˚A −2 and kD−Zr−D = 150 kcal mol −1 rad−2, respectively). Elec- trostatic interactions between ligand and dummy atoms preserve the directionality of the metal-ligand bonds and play a key role in maintaining the...
-
[3]
B. F. Hoskins and R. Robson, Infinite polymeric frame- works consisting of three dimensionally linked rod-like segments, Journal of the American Chemical Society 111, 5962–5964 (1989)
work page 1989
- [4]
-
[5]
O. M. Yaghi, G. Li, and H. Li, Selective binding and removal of guests in a microporous metal–organic frame- work, Nature 378, 703–706 (1995)
work page 1995
-
[6]
P. Z. Moghadam, A. Li, S. B. Wiggin, A. Tao, A. G. P. Maloney, P. A. Wood, S. C. Ward, and D. Fairen-Jimenez, Development of a cambridge struc- tural database subset: A collection of metal–organic frameworks for past, present, and future, Chemistry of Materials 29, 2618–2625 (2017)
work page 2017
-
[7]
A. Felix Sahayaraj, H. Joy Prabu, J. Maniraj, M. Kan- nan, M. Bharathi, P. Diwahar, and J. Salamon, Metal–organic frameworks (mofs): The next generation of materials for catalysis, gas storage, and separation, Journal of Inorganic and Organometallic Polymers and Materials 33, 1757–1781 (2023)
work page 2023
-
[8]
L. S. Andrade, H. H. Lima, C. T. Silva, W. L. Amorim, J. G. Po¸ co, A. L´ opez-Castillo, M. V. Kirillova, W. A. Carvalho, A. M. Kirillov, and D. Mandelli, Metal–organic frameworks as catalysts and biocatalysts for methane oxi- dation: The current state of the art, Coordination Chem- istry Reviews 481, 215042 (2023)
work page 2023
-
[9]
J. H. Cavka, S. Jakobsen, U. Olsbye, N. Guillou, C. Lam- berti, S. Bordiga, and K. P. Lillerud, A new zirconium inorganic building brick forming metal organic frame- works with exceptional stability, Journal of the American Chemical Society 130, 13850 (2008)
work page 2008
-
[10]
A. Dhakshinamoorthy and H. G. Andrea Santiago- Portillo, Abdullah M. Asiri, Engineering uio-66 metal organic framework for heterogeneous catalysis, Chem- CatChem 11, 899 (2018)
work page 2018
-
[11]
F. Vermoortele, B. Bueken, G. L. Bars, B. V. de Vo- orde, M. Vandichel, K. Houthoofd, A. Vimont, M. Da- turi, M. Waroquier, V. V. Speybroeck, C. Kirschhock, and D. E. D. Vos, Synthesis modulation as a tool to in- crease the catalytic activity of metal-organic frameworks: the unique case of uio-66(zr), Journal of the American Chemical Society 135, 11465 (2013)
work page 2013
-
[12]
M. Woellner, S. Hausdorf, N. Klein, P. Mueller, M. W. Smith, and S. Kaskel, Adsorption and detection of haz- ardous trace gases by metal–organic frameworks, Ad- vanced Materials 30, 1704679 (2018)
work page 2018
- [13]
-
[14]
F. Ahmadijokani, R. Mohammadkhani, S. Ahmadipouya, A. Shokrgozar, M. Rezakazemi, H. Molavi, T. M. Am- inabhavi, and M. Arjmand, Superior chemical stability of uio-66 metal-organic framework (mofs) for selective dye adsorption, Chemical Engineering Journal 399, 125346 (2020)
work page 2020
-
[15]
H. Wu, T. Yildirim, and W. Zhou, Exceptional mechan- ical stability of highly porous zirconium metal–organic framework uio-66 and its important implications, The Journal of Physical Chemistry Letters 4, 925 (2013), pMID: 26291357
work page 2013
-
[16]
P. G. Yot, K. Yang, F. Ragon, V. Dmitriev, T. Devic, P. Horcajada, C. Serre, and G. Maurin, Exploration of the mechanical behavior of metal organic frameworks uio- 66(zr) and mil-125(ti) and their nh2 functionalized ver- sions, Dalton Trans. 45, 4283 (2016)
work page 2016
-
[17]
M. R. DeStefano, T. Islamoglu, S. J. Garibay, J. T. Hupp, and O. K. Farha, Room-temperature synthesis of uio- 66 and thermal modulation of densities of defect sites, Chemistry of Materials 29, 1357–1361 (2017)
work page 2017
-
[18]
Y. Feng, Q. Chen, M. Jiang, and J. Yao, Tailoring the properties of uio-66 through defect engineering: A re- view, Industrial & Engineering Chemistry Research 58, 17646–17659 (2019)
work page 2019
-
[19]
C. S. Cox, E. Slavich, L. K. Macreadie, L. K. McKem- mish, and M. Lessio, Understanding the role of synthetic parameters in the defect engineering of uio-66: A review and meta-analysis, Chemistry of Materials35, 3057–3072 (2023)
work page 2023
- [20]
-
[21]
L. Valenzano, B. Civalleri, S. Chavan, S. Bordiga, M. H. Nilsen, S. Jakobsen, K. P. Lillerud, and C. Lamberti, Disclosing the complex structure of uio-66 metal organic framework: A synergic combination of experiment and theory, Chemistry of Materials 23, 1700–1718 (2011)
work page 2011
- [22]
-
[23]
P. Z. Moghadam, Y. G. Chung, and R. Q. Snurr, Progress toward the computational discovery of new metal–organic framework adsorbents for energy applica- tions, Nature Energy 9, 121–133 (2024)
work page 2024
-
[24]
W. Temmerman, R. Goeminne, K. S. Rawat, and V. Van Speybroeck, Computational modeling of reticu- lar materials: The past, the present, and the future, Ad- vanced Materials 37, 10.1002/adma.202412005 (2024)
-
[25]
K. Chen, S. H. Mousavi, R. Singh, R. Q. Snurr, G. Li, and P. A. Webley, Gating effect for gas adsorption in micro- porous materials—mechanisms and applications, Chem- ical Society Reviews 51, 1139–1166 (2022)
work page 2022
-
[26]
D. E. Coupry, M. A. Addicoat, and T. Heine, Exten- sion of the universal force field for metal–organic frame- works, Journal of Chemical Theory and Computation12, 5215–5225 (2016)
work page 2016
-
[27]
J. K. Bristow, D. Tiana, and A. Walsh, Transferable force field for metal–organic frameworks from first-principles: Btw-ff, Journal of Chemical Theory and Computation 10, 4644–4652 (2014)
work page 2014
-
[28]
S. Bureekaew, S. Amirjalayer, M. Tafipolsky, C. Spick- ermann, T. K. Roy, and R. Schmid, Mof-ff – a flexi- ble first-principles derived force field for metal-organic frameworks, physica status solidi (b) 250, 1128–1141 10 (2013)
work page 2013
-
[29]
L. Vanduyfhuys, S. Vandenbrande, T. Verstraelen, R. Schmid, M. Waroquier, and V. Van Speybroeck, Quickff: A program for a quick and easy derivation of force fields for metal-organic frameworks from ab initio input, Journal of Computational Chemistry 36, 1015–1027 (2015)
work page 2015
-
[30]
J. Heinen and D. Dubbeldam, On flexible force fields for metal–organic frameworks: Recent developments and fu- ture prospects, WIREs Computational Molecular Science 8, 10.1002/wcms.1363 (2018)
-
[31]
P. G. Boyd, S. M. Moosavi, M. Witman, and B. Smit, Force-field prediction of materials properties in metal- organic frameworks, The Journal of Physical Chemistry Letters 8, 357 (2017), pMID: 28008758
work page 2017
-
[32]
P. Dobbelaere, S. Vandenhaute, and V. Van Spey- broeck, Cluster-based machine learning potentials to de- scribe disordered metal–organic frameworks up to the mesoscale, Chemistry of Materials 37, 5696–5709 (2025)
work page 2025
-
[33]
O. Tayfuroglu and S. Keskin, Transforming mof modeling with machine-learned potentials: Progress and perspec- tives, Journal of Chemical Information and Modeling 66, 1964–1981 (2026)
work page 1964
-
[34]
T. P. Senftle, S. Hong, M. M. Islam, S. B. Kylasa, Y. Zheng, Y. K. Shin, C. Junkermeier, R. Engel-Herbert, M. J. Janik, H. M. Aktulga, T. Verstraelen, A. Grama, and A. C. T. van Duin, The reaxff reactive force-field: de- velopment, applications and future directions, npj Com- putational Materials 2, 10.1038/npjcompumats.2015.11 (2016)
-
[35]
S. Zhuo, Y. Wang, K. Qi, Y. Sun, Y. Zhou, J. Wang, and Z. Li, Simulating the self-assembly of mof-5 using reactive force field (reaxff), The Journal of Physical Chemistry C 10.1021/acs.jpcc.5c08014 (2026)
-
[36]
N. Castel and F.-X. Coudert, Challenges in molecular dynamics of amorphous ZIFs using reactive force fields, The Journal of Physical Chemistry C 126, 19532 (2022)
work page 2022
-
[37]
N. Castel and F.-X. Coudert, Computation of finite tem- perature mechanical properties of zeolitic imidazolate framework glasses by molecular dynamics, Chemistry of Materials 35, 4038 (2023)
work page 2023
-
[38]
D. Biswal and P. G. Kusalik, Probing molecular mecha- nisms of self-assembly in metal–organic frameworks, ACS Nano 11, 258 (2016)
work page 2016
-
[39]
S. R. G. Balestra and R. Semino, Computer simulation of the early stages of self-assembly and thermal decom- position of ZIF-8, The Journal of Chemical Physics 157, 184502 (2022)
work page 2022
-
[40]
D. Biswal and P. G. Kusalik, Molecular simulations of self-assembly processes in metal-organic frameworks: Model dependence, J. Chem. Phys. 147, 044702 (2017)
work page 2017
-
[41]
S. A. Gargari and R. Semino, Unveiling zif-8 nucleation mechanisms through molecular simulation: Role of tem- perature, solvent, and reactant concentration, Chemistry of Materials 37, 9460–9470 (2025)
work page 2025
-
[42]
E. M´ endez and R. Semino, Microscopic mechanism of thermal amorphization of zif-4 and melting of zif-zni revealed via molecular dynamics and machine learn- ing techniques, Journal of Materials Chemistry A 12, 4572–4582 (2024)
work page 2024
-
[43]
E. M´ endez and R. Semino, Phase diagram of zif-4 from computer simulations, Journal of Materials Chemistry A 12, 31108–31115 (2024)
work page 2024
-
[44]
E. M´ endez and R. Semino, Thermodynamic insights into the self-assembly of zeolitic imidazolate frameworks from computer simulations, Chem. Sci. 16, 11979 (2025)
work page 2025
-
[45]
S. Jawahery, N. Rampal, S. M. Moosavi, M. Wit- man, and B. Smit, Ab initio flexible force field for metal–organic frameworks using dummy model coordi- nation bonds, Journal of Chemical Theory and Compu- tation 15, 3666–3677 (2019)
work page 2019
- [46]
-
[47]
D. Golo, M. S. G. Ahlquist, and H. Su, Development and application of fe3+, al3+, cr3+ dummy atom models for metal–organic frameworks, ACS Omega 10, 3801–3807 (2025)
work page 2025
-
[48]
A. P. Thompson, H. M. Aktulga, R. Berger, D. S. Bolin- tineanu, W. M. Brown, P. S. Crozier, P. J. in 't Veld, A. Kohlmeyer, S. G. Moore, T. D. Nguyen, R. Shan, M. J. Stevens, J. Tranchida, C. Trott, and S. J. Plimpton, LAMMPS - a flexible simulation tool for particle-based materials modeling at the atomic, meso, and continuum scales, Computer Physics Comm...
work page 2022
-
[49]
Q. Yang, H. Jobic, F. Salles, D. Kolokolov, V. Guillerm, C. Serre, and G. Maurin, Probing the dynamics of co2 and ch4 within the porous zirconium terephthalate uio- 66(zr): A synergic combination of neutron scattering measurements and molecular simulations, Chemistry–A European Journal 17, 8882 (2011)
work page 2011
-
[50]
S. M. J. Rogge, J. Wieme, L. Vanduyfhuys, S. Vanden- brande, G. Maurin, T. Verstraelen, M. Waroquier, and V. Van Speybroeck, Thermodynamic insight in the high- pressure behavior of uio-66: Effect of linker defects and linker expansion, Chemistry of Materials28, 5721 (2016), pMID: 27594765
work page 2016
-
[51]
A. Schaate, P. Roy, A. Godt, J. Lippke, F. Waltz, M. Wiebcke, and P. Behrens, Modulated synthesis of zr-based metal–organic frameworks: From nano to sin- gle crystals, Chemistry – A European Journal 17, 6643 (2011)
work page 2011
-
[52]
Y. Zhao, Q. Zhang, Y. Li, R. Zhang, and G. Lu, Large- scale synthesis of monodisperse uio-66 crystals with tunable sizes and missing linker defects via acid/base co-modulation, ACS Applied Materials & Interfaces 9, 15079 (2017), pMID: 28425280
work page 2017
-
[53]
N. Hosadoddi Srikantamurthy, J. F. Olorunyomi, C. M. Doherty, P. C. Sherrell, and X. Mulet, Aqueous synthesis of uio-66 metal-organic frameworks with enhanced crys- tallinity and surface area, Advanced Sustainable Systems 9, e00854 (2025)
work page 2025
-
[54]
M. Chalaris and J. Samios, Systematic molecular dynam- ics studies of liquid n, n-dimethylformamide using opti- mized rigid force fields: Investigation of the thermody- namic, structural, transport and dynamic properties, The Journal of Chemical Physics 112, 8581–8594 (2000)
work page 2000
-
[55]
A. D. C. Morales, I. G. Economou, C. J. Peters, and J. I. Siepmann, Influence of simulation protocols on the effi- ciency of gibbs ensemble monte carlo simulations, Molec- ular Simulation 39, 1135 (2013)
work page 2013
-
[56]
D. Frenkel and B. Smit, Understanding molecular simu- lation: from algorithms to applications (Elsevier, 2023)
work page 2023
-
[57]
S. Nos´ e, A unified formulation of the constant tempera- ture molecular dynamics methods, The Journal of Chem- 11 ical Physics 81, 511 (1984)
work page 1984
-
[58]
W. G. Hoover, Canonical dynamics: Equilibrium phase-space distributions, Physical Review A 31, 10.1103/PhysRevA.31.1695 (1985)
-
[59]
A. K. Rappe, C. J. Casewit, K. S. Colwell, W. A. I. God- dard, and W. M. Skiff, Uff, a full periodic table force field for molecular mechanics and molecular dynamics simu- lations, Journal of the American Chemical Society 114, 10024 (1992)
work page 1992
-
[60]
M. Kaur, Stress-strain analysis of cubic crystals, in Fu- turistic Trends in Physical Sciences , IIP Series, Vol. 3 (Iterative International Publishers (IIP), 2023) pp. 137– 149
work page 2023
-
[61]
A. Laio and M. Parrinello, Escaping free-energy min- ima, Proceedings of the National Academy of Sciences 99, 12562–12566 (2002)
work page 2002
-
[62]
A. Barducci, G. Bussi, and M. Parrinello, Well-tempered metadynamics: A smoothly converging and tunable free- energy method, Physical Review Letters 100, 020603 (2008)
work page 2008
-
[63]
G. A. Tribello, M. Bonomi, D. Branduardi, C. Camil- loni, and G. Bussi, Plumed 2: New feathers for an old bird, Computer Physics Communications 185, 604–613 (2014). 12 Supporting Information Partially reactive force field for the UiO-66 metal-organic framework Akanksha Nawani, Rocio Semino Sorbonne Universit´ e, CNRS, Physico-chimie des Electrolytes et Na...
work page 2014
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.