Bipartite Cholesky Graph Networks for Many-Body Quantum Chemistry
Pith reviewed 2026-06-29 23:22 UTC · model grok-4.3
The pith
Cholesky factorization of the ERI tensor induces a bipartite graph network that predicts molecular correlation energies by treating orbitals and auxiliary nodes as separate sets.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
Tensor factorization of the ERI via density-fitted Cholesky decomposition naturally induces a bipartite message-passing architecture in which orbital nodes and auxiliary interaction nodes form distinct partitions; message passing between these partitions preserves the original interaction topology at reduced O(N^3) complexity and produces more accurate correlation-energy predictions than compressed-integral baselines on the tested diatomic set.
What carries the argument
The bipartite graph network whose two partitions are orbital degrees of freedom and auxiliary interaction nodes obtained from the density-fitted Cholesky decomposition of the ERI tensor.
If this is right
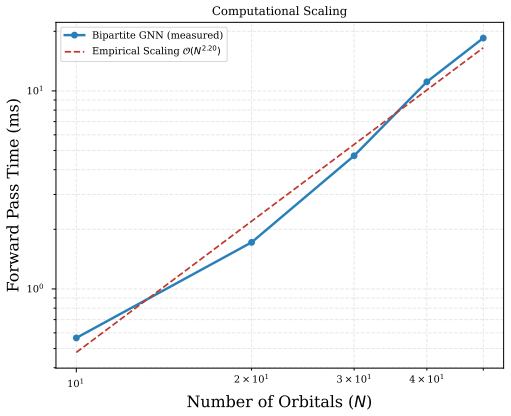
- The model maintains interaction topology at O(N^3) cost instead of the conventional O(N^4) ERI scaling.
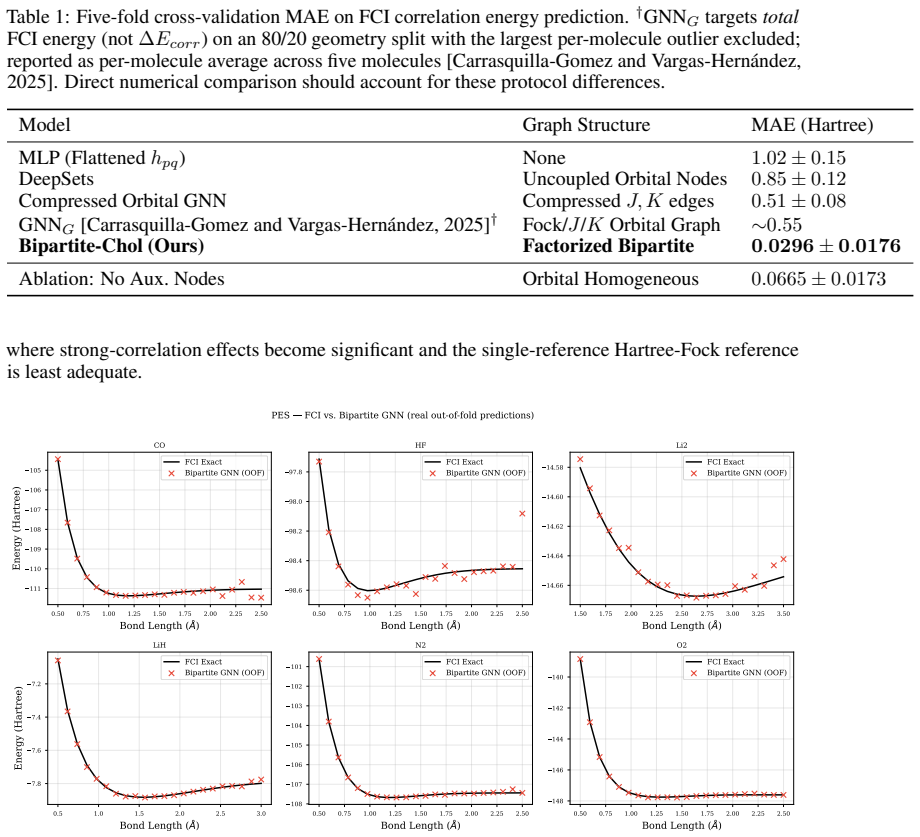
- In-distribution accuracy reaches 0.0296 Ha MAE on the diatomic test set under five-fold cross-validation.
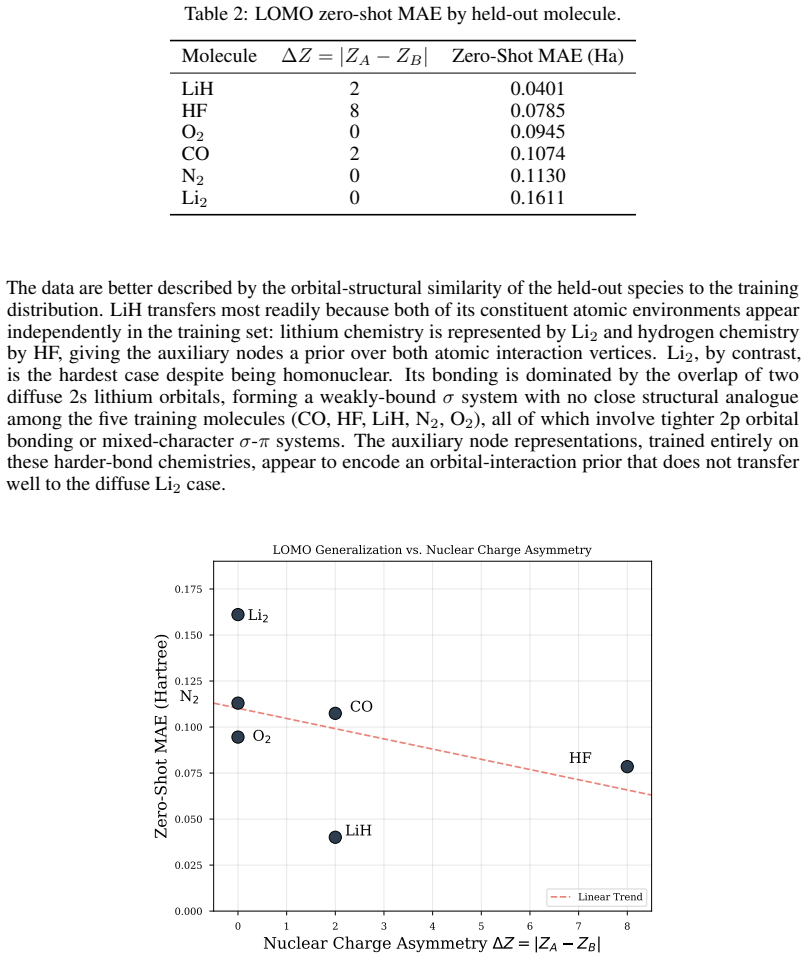
- Zero-shot transfer error varies by nearly a factor of four across molecules and tracks orbital-environment similarity rather than nuclear-charge asymmetry.
- The architecture supplies a concrete route to keep higher-order electron-correlation structure inside graph networks without full tensor storage.
Where Pith is reading between the lines
- The same bipartite construction could be applied to other tensor factorizations that separate orbital and auxiliary indices, such as density fitting without Cholesky.
- If the orbital-environment similarity metric proves predictive, training-set curation could be guided by orbital-space overlap rather than by molecular identity alone.
- Extension to polyatomic systems would test whether the O(N^3) scaling and bipartite separation remain advantageous once three-center integrals involve more than two centers.
Load-bearing premise
That the Cholesky factorization of the ERI tensor automatically creates a bipartite message-passing structure that retains higher-order interaction information more effectively than any compressed orbital representation.
What would settle it
A controlled experiment in which the same ERI data are fed to an otherwise identical graph network that collapses the auxiliary factors into scalar node features and still achieves equal or lower MAE on the identical 132-geometry diatomic benchmark.
Figures

read the original abstract
Accurate prediction of molecular correlation energies from first principles requires resolving the {O}(N^4) electron repulsion integral (ERI) tensor. Existing graph neural network approaches to the electronic structure problem often compress this tensor into low-rank scalar features, discarding higher-order interaction structures relevant to electron correlation. In this work, we demonstrate that tensor factorization of the ERI naturally induces a structured bipartite message-passing architecture that preserves access to higher-order interaction structure more effectively than compressed orbital representations. By utilizing the density-fitted Cholesky decomposition of the ERI tensor, we derive a bipartite graph network that models orbital degrees of freedom and auxiliary interaction nodes as distinct sets, maintaining interaction topology at a reduced theoretical complexity of {O}(N^3). Evaluated on 132 geometries of six diatomic molecules with Full Configuration Interaction (FCI) reference energies, our factorized representation achieves an in-distribution Mean Absolute Error (MAE) of 0.0296 Ha under five-fold cross-validation, a substantial improvement over compressed-integral baselines. Leave-one-molecule-out validation reveals that zero-shot generalization varies by nearly a factor of four across molecular species and correlates with the structural similarity of the held-out molecule's orbital environment to the training distribution, rather than with nuclear charge asymmetry alone.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The paper claims that density-fitted Cholesky decomposition of the ERI tensor naturally induces a bipartite graph neural network architecture with orbital nodes and auxiliary interaction nodes as distinct sets. This maintains interaction topology at O(N^3) theoretical complexity while preserving higher-order structures better than compressed orbital representations. On 132 geometries of six diatomic molecules with FCI references, the model achieves 0.0296 Ha MAE under five-fold cross-validation (substantial improvement over baselines); leave-one-molecule-out generalization varies by a factor of ~4 and correlates with orbital-environment similarity to the training set rather than nuclear charge.
Significance. If the central architectural claim holds beyond the current test set, the work would provide a concrete route to embed tensor-factorized ERI structure directly into message-passing networks, offering a principled alternative to scalar compression for correlation-energy models. The O(N^3) scaling and explicit bipartite topology are attractive for scaling to larger orbital spaces. However, the restriction to diatomics means the significance for genuine many-body quantum chemistry remains provisional; broader validation on polyatomics would be required to establish whether the factorization-induced bipartiteness confers a genuine advantage over other low-rank or compressed-integral approaches.
major comments (2)
- [Abstract] Abstract (evaluation paragraph): The central claim that the bipartite Cholesky architecture 'preserves access to higher-order interaction structure more effectively than compressed orbital representations' is supported solely by results on 132 geometries of six diatomic molecules. Diatomics possess low-dimensional orbital spaces and limited many-body character; the leave-one-molecule-out variation (factor of ~4, correlated with orbital-environment similarity) is consistent with the model fitting local patterns rather than demonstrating general access to higher-order tensor structure. This dataset choice is load-bearing for the many-body claim and leaves the architectural advantage untested on systems where higher-order ERI contributions are prominent.
- [Abstract] Abstract (numerical claim): The reported in-distribution MAE of 0.0296 Ha under five-fold cross-validation is presented without accompanying information on baseline definitions, cross-validation splits, error bars, or statistical significance of the improvement over compressed-integral baselines. Because the abstract states this numerical result as the primary evidence for the factorized representation's superiority, the absence of these details prevents verification of the central performance claim.
Simulated Author's Rebuttal
We thank the referee for the constructive feedback. We address each major comment below, indicating planned revisions where appropriate.
read point-by-point responses
-
Referee: [Abstract] Abstract (evaluation paragraph): The central claim that the bipartite Cholesky architecture 'preserves access to higher-order interaction structure more effectively than compressed orbital representations' is supported solely by results on 132 geometries of six diatomic molecules. Diatomics possess low-dimensional orbital spaces and limited many-body character; the leave-one-molecule-out variation (factor of ~4, correlated with orbital-environment similarity) is consistent with the model fitting local patterns rather than demonstrating general access to higher-order tensor structure. This dataset choice is load-bearing for the many-body claim and leaves the architectural advantage untested on systems where higher-order ERI contributions are prominent.
Authors: We agree that the current results are limited to diatomic molecules and that this restricts the strength of claims about advantages for higher-order many-body interactions in more complex systems. The observed variation in leave-one-molecule-out performance is indeed consistent with sensitivity to orbital-environment similarity. We will revise the abstract and relevant discussion sections to qualify the scope of the architectural claims, explicitly noting the provisional nature of the many-body advantage and the need for future validation on polyatomics. revision: yes
-
Referee: [Abstract] Abstract (numerical claim): The reported in-distribution MAE of 0.0296 Ha under five-fold cross-validation is presented without accompanying information on baseline definitions, cross-validation splits, error bars, or statistical significance of the improvement over compressed-integral baselines. Because the abstract states this numerical result as the primary evidence for the factorized representation's superiority, the absence of these details prevents verification of the central performance claim.
Authors: Full details on baseline definitions, cross-validation splits, and complete results (including error bars) appear in the Methods and Results sections. We acknowledge that the abstract would be strengthened by brief additional context on these elements. We will revise the abstract to reference the baselines more explicitly and note that statistical details are provided in the main text. revision: yes
Circularity Check
No significant circularity; derivation self-contained against external benchmarks
full rationale
The paper constructs the bipartite graph network directly from the structure of the density-fitted Cholesky decomposition of the ERI tensor, treating orbital and auxiliary nodes as distinct sets whose topology follows from the factorization. This is presented as an architectural choice motivated by tensor properties rather than any fitted parameter or self-referential definition. Evaluation relies on independent FCI reference energies for the reported MAE under cross-validation, with no equations or steps shown that reduce a prediction to its own inputs by construction. No load-bearing self-citations, uniqueness theorems, or ansatzes imported from prior author work are invoked in the provided text. The central claim therefore remains independent of the reported results.
Axiom & Free-Parameter Ledger
axioms (1)
- domain assumption Density-fitted Cholesky decomposition of the ERI tensor preserves higher-order interaction structure sufficiently for bipartite message passing.
Reference graph
Works this paper leans on
-
[1]
PennyLane: Automatic differentiation of hybrid quantum-classical computations
doi: 10.1002/qua.560120408. Ville Bergholm, Josh Izaac, Maria Schuld, Christian Gogolin, Shahnawaz Ahmed, Vishnu Ajith, M Sohaib Alam, Guillermo Alonso-Linaje, B AkashNarayanan, Ali Asadi, et al. PennyLane: Auto- matic differentiation of hybrid quantum-classical computations.arXiv preprint arXiv:1811.04968,
work page internal anchor Pith review Pith/arXiv arXiv doi:10.1002/qua.560120408
-
[2]
Juan Carrasquilla-Gomez and Rodrigo A
doi: 10.1063/5.0076588. Juan Carrasquilla-Gomez and Rodrigo A. Vargas-Hernández. Graph neural networks on one- and two- body integrals for molecular energy prediction. InMachine Learning and the Physical Sciences Workshop, NeurIPS 2025,
-
[3]
Valerii Chuiko and Paul W Ayers
doi: 10.1063/1.5126701. Valerii Chuiko and Paul W Ayers. Predicting energy of the quantum system from one- and two- electron integrals using deep learning.arXiv preprint arXiv:2504.03849,
-
[4]
Justin Gilmer, Samuel S Schoenholz, Patrick F Riley, Oriol Vinyals, and George E Dahl
doi: 10.48550/ arXiv.2504.03849. Justin Gilmer, Samuel S Schoenholz, Patrick F Riley, Oriol Vinyals, and George E Dahl. Neural message passing for quantum chemistry. InInternational conference on machine learning, pages 1263–1272. PMLR,
-
[5]
Thomas Bondo Pedersen, Francesco Aquilante, and Roland Lindh
doi: 10.1063/1.1578621. Thomas Bondo Pedersen, Francesco Aquilante, and Roland Lindh. Density fitting with auxiliary basis sets from Cholesky decompositions.Theoretical Chemistry Accounts, 124(1):1–10,
-
[6]
Zhuoran Qiao, Matthew Welborn, Animashree Anandkumar, Frederick R Manby, and Thomas F Miller III
doi: 10.1007/s00214-009-0608-y. Zhuoran Qiao, Matthew Welborn, Animashree Anandkumar, Frederick R Manby, and Thomas F Miller III. Orbnet: Deep learning for quantum chemistry using symmetry-adapted atomic-orbital features.The Journal of Chemical Physics, 153(12):124111,
-
[7]
doi: 10.1063/5.0021955. Raghunathan Ramakrishnan, Pavlo O Dral, Matthias Rupp, and O Anatole von Lilienfeld. Big data meets quantum chemistry approximations: The δ-machine learning approach.Journal of Chemical Theory and Computation, 11(5):2087–2096,
-
[8]
doi: 10.1021/acs.jctc.5b00099. Kristof T Schütt, Huziel E Sauceda, Pieter-Jan Kindermans, Alexandre Tkatchenko, and Klaus- Robert Müller. Schnet: A continuous-filter convolutional neural network for modeling quantum interactions.The Journal of Chemical Physics, 148(24):241722,
-
[9]
u tt, H. E. Sauceda, P.-J. Kindermans, A. Tkatchenko, and K.-R. M \
doi: 10.1063/1.5019779. Kristof T Schütt, Michael Gastegger, Alexandre Tkatchenko, Klaus-Robert Müller, and Reinhard J Maurer. Unifying machine learning and quantum chemistry with a deep neural network for molecu- lar wavefunctions.Nature Communications, 10(1):5024,
-
[10]
doi: 10.1038/s41467-019-12875-2. Matthew Welborn, Lixue Cheng, and Thomas F Miller III. Transferability in machine learning for electronic structure via the molecular orbital basis.Journal of Chemical Theory and Computation, 14(9):4772–4779,
-
[11]
Shuo Zhang, Yang Liu, and Lei Xie
doi: 10.1021/acs.jctc.8b00636. Shuo Zhang, Yang Liu, and Lei Xie. A universal framework for accurate and efficient ge- ometric deep learning of molecular systems.Scientific Reports, 13(1):19171,
-
[12]
doi: 10.1038/s41598-023-46382-8. 9 A Extended Mathematical Formulation This appendix details the theoretical mappings connecting the quantum many-body Hamiltonian to the factorized bipartite graph architecture, as well as the formal tensor operations comprising the message-passing framework. A.1 The Coulomb Metric and Positive Semi-Definiteness The non-re...
-
[13]
Because linear dependencies in the auxiliary basis scale approximately as Naux ∼N in practice [Koch et al., 2003], the theoretical scaling is broadly bounded byO(N 3)
to O(N 2Naux). Because linear dependencies in the auxiliary basis scale approximately as Naux ∼N in practice [Koch et al., 2003], the theoretical scaling is broadly bounded byO(N 3). A.3 Tensor Algebra of Bipartite Message Passing Let x(t) p ∈R H be the latent representation of orbital node p at layer t, and h(t) L ∈R H the representa- tion of auxiliary n...
2003
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.