Bridging the NISQ and Fault-Tolerant Regimes: Generative-ML-Assisted Quantum Selected CI for Molecular Simulations
Pith reviewed 2026-06-30 05:58 UTC · model grok-4.3
The pith
Integrating a linear-scaling ansatz and a generative machine learning model into quantum-selected configuration interaction cuts the classical resources needed for molecular binding energy calculations.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
The authors claim that integrating LCNot-UCCSD into the QSCI framework and employing QSCI-RBM for generative modeling allows accurate electronic structure calculations on NISQ-era simulators for protein-ligand systems, achieving this with substantially reduced classical computational overhead compared to existing methods.
What carries the argument
The LCNot-UCCSD ansatz providing O(N^4) MP2-based initialization for the QSCI procedure, together with the RBM acting as a compact generative model for subspace expansion in place of traditional recovery methods.
Load-bearing premise
The assumption that performance on an ideal state-vector simulator with artificial error levels will carry over to real NISQ hardware without significant additional costs from error mitigation or increased circuit depths.
What would settle it
Running the QSCI-RBM workflow on physical quantum processors for one of the tested molecules and measuring whether the resource savings and accuracy are maintained under real noise conditions.
Figures






read the original abstract
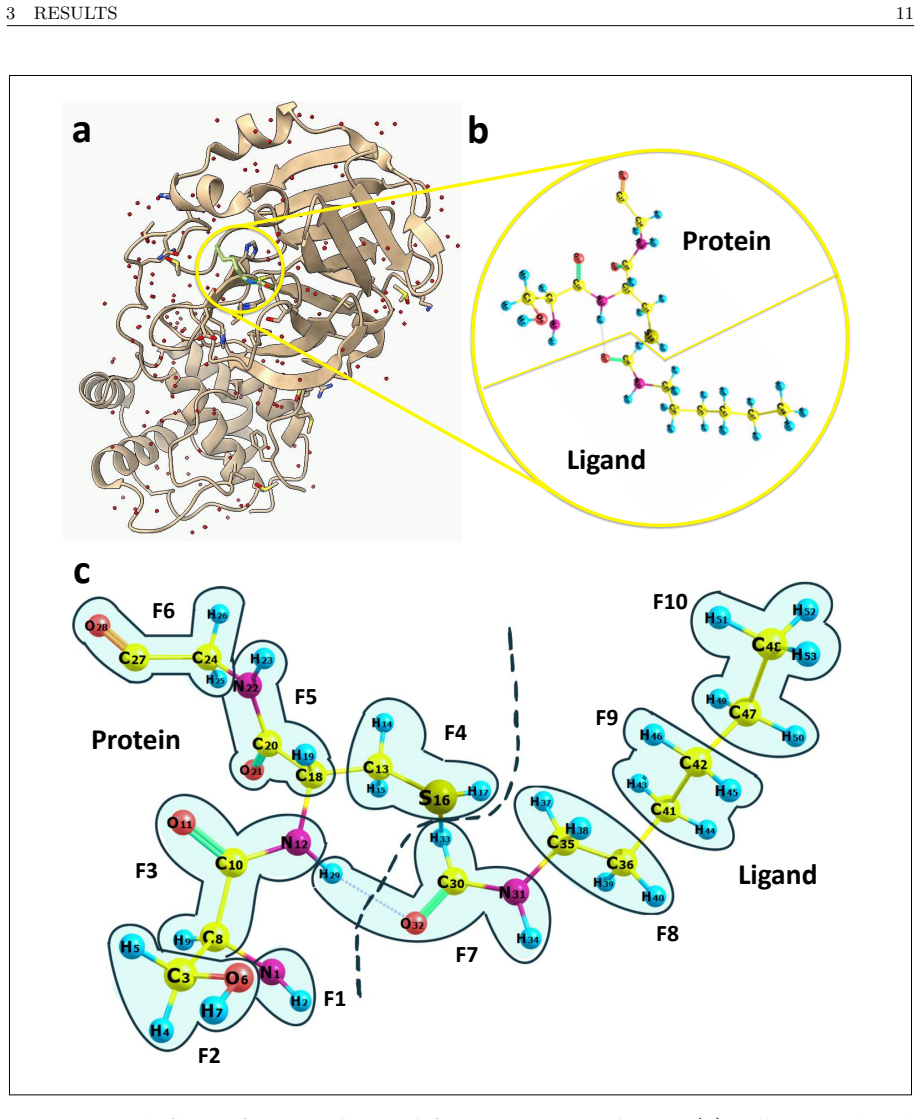
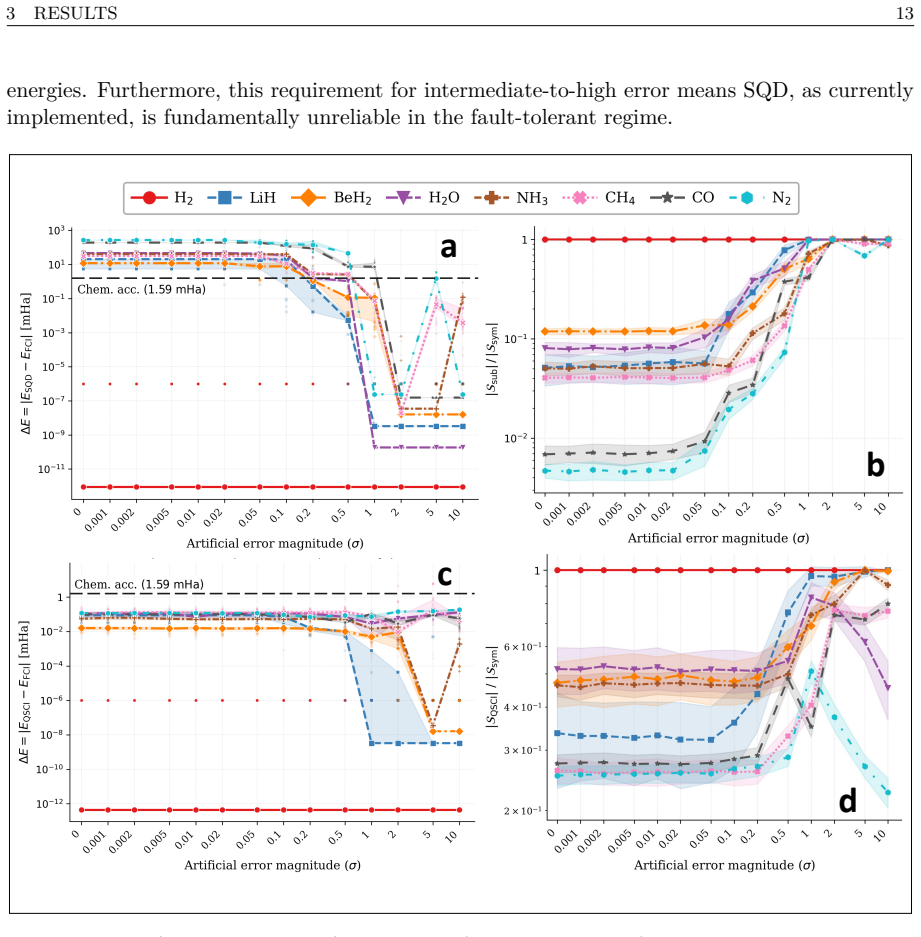
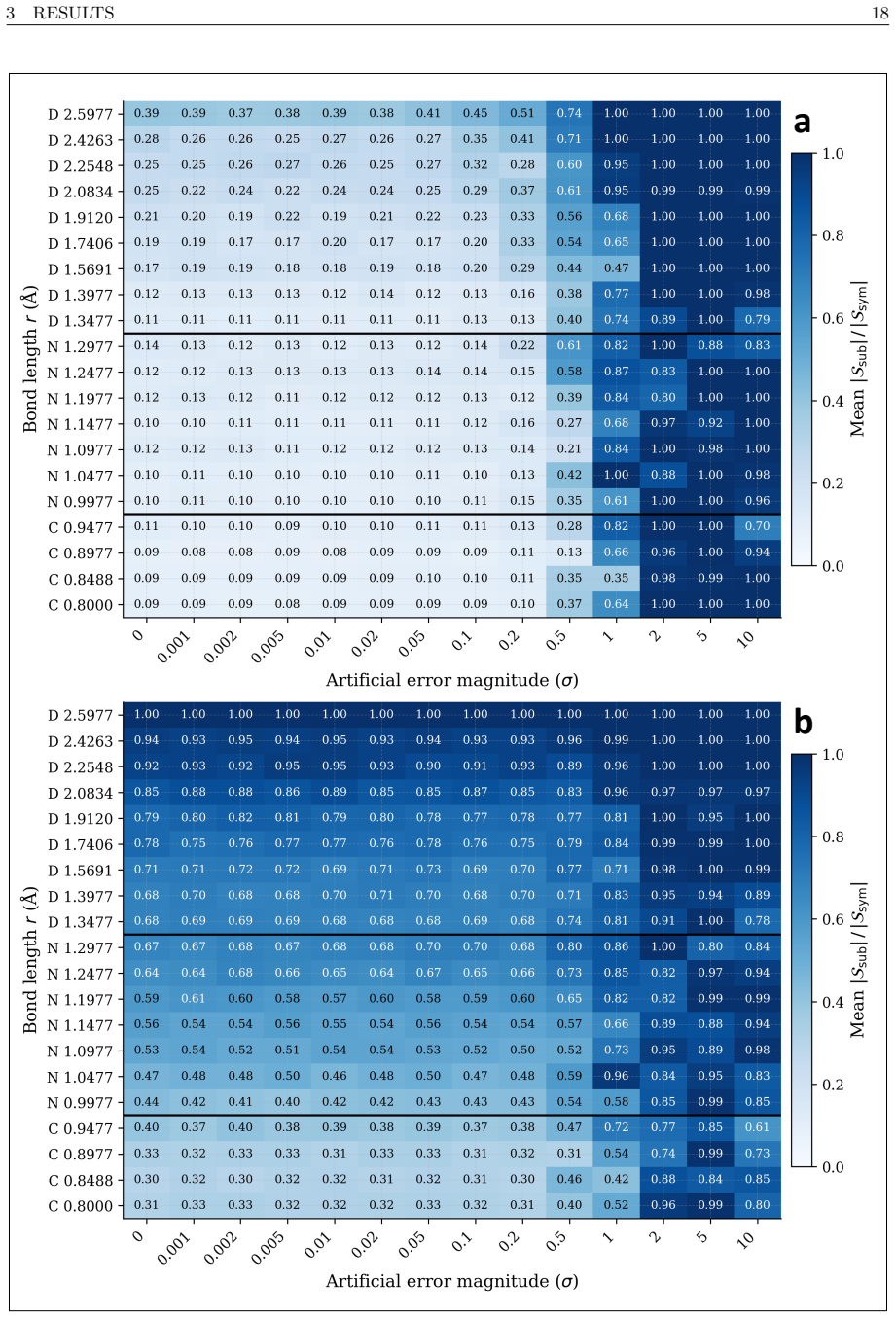
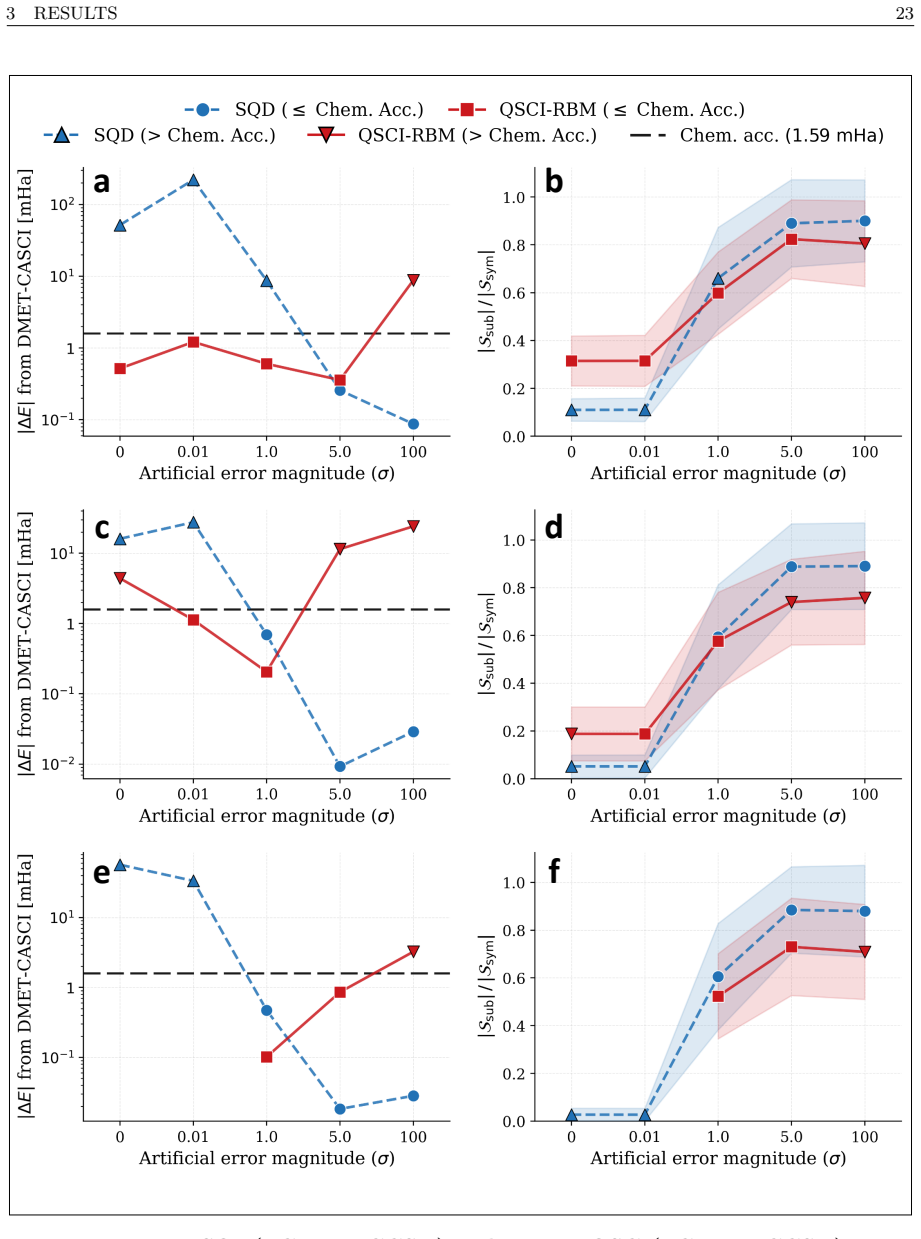
Calculation of binding energies for protein-ligand molecular systems requires accurate treatment of the electronic structure, a quantum chemistry problem that scales exponentially on classical hardware, while current quantum hardware remains too noisy for the required circuit depths. This report presents a hybrid quantum-classical workflow performed on the Fujitsu FX700 ideal state-vector simulator using QARP that addresses two structural inefficiencies in quantum-sampling-based diagonalization workflows. First, we integrate the Linear Scaling CNOT UCCSD (LCNot-UCCSD) ansatz into the QSCI framework, replacing the $\mathcal{O}(N^6)$ CCSD parameter initialization of the competing LUCJ ansatz approach with $\mathcal{O}(N^4)$ MP2-amplitude initialization. Second, we introduce QSCI-RBM, a variant that replaces the configuration recovery of the SQD framework with a Restricted Boltzmann Machine (RBM) acting as a compact generative subspace expansion model. Both are evaluated on eight different molecules in STO-3G across 14 controlled artificial error levels with 100 independent runs each, validated on potential energy surface scans of the N$_2$ molecule in cc-pVDZ, and embedded within DMET to treat the FDA-approved antiviral Amantadine (C$_{10}$H$_{17}$N, 11 DMET fragments) and the active region of the SARS-CoV-2 main protease complexed with its covalent inhibitor Carmofur (PDB: 7BUY, C$_{15}$H$_{28}$N$_4$O$_5$S, 10 fragments). To our knowledge, this is the first deployment of LCNot-UCCSD within QSCI on a quantum computing simulator, and the first DMET-QSCI(LCNot-UCCSD)-RBM application to an industry-relevant protein-ligand system. By utilizing a fraction of the classical computing resources required by the current state-of-the-art work by Cleveland Clinic, RIKEN, and IBM Quantum, this approach enables more efficient and economical drug discovery simulations for the industry.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The paper presents a hybrid quantum-classical workflow for molecular electronic structure calculations that integrates the LCNot-UCCSD ansatz into the QSCI framework (replacing O(N^6) CCSD initialization with O(N^4) MP2 amplitudes) and introduces QSCI-RBM, which uses a Restricted Boltzmann Machine for configuration recovery in place of SQD methods. The approach is evaluated on an ideal state-vector simulator for eight molecules in STO-3G across 14 artificial error levels (100 runs each), N2 PES scans in cc-pVDZ, and DMET embeddings of two protein-ligand systems (Amantadine and the SARS-CoV-2 main protease with Carmofur), with the central claim being that it uses a fraction of the classical resources required by prior Cleveland Clinic/RIKEN/IBM work.
Significance. If the unshown numerical results and error metrics support the efficiency claims, the combination of a cheaper ansatz initialization and generative-model subspace expansion could reduce the classical overhead in quantum-selected CI workflows and extend their reach to larger embedded systems relevant to drug discovery. The work also supplies the first reported use of LCNot-UCCSD inside QSCI and the first DMET-QSCI-RBM application to an industry protein-ligand complex.
major comments (2)
- [Abstract] Abstract: The evaluation protocol (8 molecules, 14 error levels, 100 runs, PES scans, DMET on two systems) is described in detail, yet the manuscript supplies no numerical results, error bars, timing data, or comparison tables. Without these data the headline claim that the method uses only a fraction of the classical resources of the Cleveland Clinic/RIKEN/IBM reference cannot be assessed.
- [Abstract] Abstract and evaluation description: All reported runs are performed on the Fujitsu FX700 ideal state-vector simulator with controlled artificial error levels. No data or analysis is given on circuit-depth overhead, real-device noise, or additional error-mitigation costs that would appear on actual NISQ hardware; if these costs exceed the simulated savings, both the resource-comparison claim and the drug-discovery applicability statement fail.
Simulated Author's Rebuttal
We thank the referee for their careful reading and for highlighting these important points regarding the presentation of results and the scope of the simulations. We address each comment below and commit to revisions that strengthen the manuscript without overstating its current content.
read point-by-point responses
-
Referee: [Abstract] Abstract: The evaluation protocol (8 molecules, 14 error levels, 100 runs, PES scans, DMET on two systems) is described in detail, yet the manuscript supplies no numerical results, error bars, timing data, or comparison tables. Without these data the headline claim that the method uses only a fraction of the classical resources of the Cleveland Clinic/RIKEN/IBM reference cannot be assessed.
Authors: We agree that the current manuscript version does not present the numerical results, error bars, timing data, or comparison tables needed to substantiate the resource-efficiency claim. This omission prevents independent assessment of the headline statement. In the revised manuscript we will add the full set of simulation outcomes (including per-molecule error metrics with standard deviations from the 100 runs, wall-clock timings, and direct resource comparisons) in the Results section and will insert a concise quantitative summary into the abstract. revision: yes
-
Referee: [Abstract] Abstract and evaluation description: All reported runs are performed on the Fujitsu FX700 ideal state-vector simulator with controlled artificial error levels. No data or analysis is given on circuit-depth overhead, real-device noise, or additional error-mitigation costs that would appear on actual NISQ hardware; if these costs exceed the simulated savings, both the resource-comparison claim and the drug-discovery applicability statement fail.
Authors: The evaluations were intentionally restricted to an ideal state-vector simulator to isolate the algorithmic contributions of LCNot-UCCSD initialization and RBM-based recovery. We will add a dedicated subsection that reports the circuit depths required by the LCNot-UCCSD ansatz, estimates the two-qubit gate counts, and discusses how standard error-mitigation techniques (e.g., zero-noise extrapolation or probabilistic error cancellation) could be combined with the workflow. We will also qualify the drug-discovery applicability statement to reflect that real-hardware overheads remain to be quantified experimentally. revision: partial
Circularity Check
No significant circularity; derivations introduce independent components without reduction to inputs or self-citations.
full rationale
The paper defines LCNot-UCCSD via MP2 amplitudes (O(N^4)) replacing CCSD initialization and introduces QSCI-RBM as a generative model for subspace expansion, both presented as novel integrations into the QSCI framework. These steps are evaluated via simulator runs but do not reduce by construction to fitted parameters renamed as predictions, self-definitions, or load-bearing self-citations. The resource-efficiency claim rests on explicit comparisons to external prior work rather than internal tautologies, leaving the derivation chain self-contained against external benchmarks.
Axiom & Free-Parameter Ledger
Reference graph
Works this paper leans on
-
[1]
Ligand-binding affinity estimates supported by quantum- mechanical methods,
U. Ryde and P. Söderhjelm, “Ligand-binding affinity estimates supported by quantum- mechanical methods,”Chemical Reviews, vol. 116, no. 9, pp. 5520–5566, Apr 2016. [Online]. Available: https://doi.org/10.1021/acs.chemrev.5b00630
-
[2]
A. Pecina, J. Fanfrlík, M. Lepšík, and J. Řezáč, “Sqm2.20: Semiempirical quantum- mechanical scoring function yields dft-quality protein–ligand binding affinity predictions in minutes,”Nature Communications, vol. 15, no. 1, p. 1127, Feb 2024. [Online]. Available: https://doi.org/10.1038/s41467-024-45431-8
-
[3]
Distributed implementation of full configuration interaction for one trillion determinants,
H. Gao, S. Imamura, A. Kasagi, and E. Yoshida, “Distributed implementation of full configuration interaction for one trillion determinants,”Journal of Chemical Theory and Computation, vol. 20, no. 3, pp. 1185–1192, Feb 2024. [Online]. Available: https://doi.org/10.1021/acs.jctc.3c01190
-
[4]
Numerically exact configuration interaction at quadrillion-determinant scale,
A. Shayit, C. Liao, S. Upadhyay, H. Hu, T. Zhang, A. E. DePrince III, C. Yang, and X. Li, “Numerically exact configuration interaction at quadrillion-determinant scale,”Nature Communications, vol. 16, no. 1, p. 11016, Dec 2025. [Online]. Available: https://doi.org/10.1038/s41467-025-65967-7
-
[5]
Szabo and N
A. Szabo and N. S. Ostlund,Modern Quantum Chemistry: Introduction to Advanced Electronic Structure Theory, 1st ed., ser. Dover Books on Chemistry. Mineola, NY: Dover Publications, 1996. [Online]. Available: https://books.google.co.in/books?id= k-DcCgAAQBAJ
1996
-
[6]
T. Helgaker, P. Jørgensen, and J. Olsen,Molecular Electronic-Structure Theory, 1st ed. John Wiley & Sons, Ltd, 2000. [Online]. Available: https://onlinelibrary.wiley.com/doi/abs/ 10.1002/9781119019572.ch1
-
[7]
Insights into current limitations of density functional theory,
A. J. Cohen, P. Mori-Sánchez, and W. Yang, “Insights into current limitations of density functional theory,”Science, vol. 321, no. 5890, pp. 792–794, Aug 2008. [Online]. Available: https://www.science.org/doi/abs/10.1126/science.1158722
-
[8]
Challenges for density functional theory,
——, “Challenges for density functional theory,”Chemical Reviews, vol. 112, no. 1, pp. 289–320, Dec 2012. [Online]. Available: https://doi.org/10.1021/cr200107z
-
[9]
K. Kanno, M. Kohda, R. Imai, S. Koh, K. Mitarai, W. Mizukami, and Y. O. Nakagawa, “Quantum-selected configuration interaction: classical diagonalization of hamiltonians in subspaces selected by quantum computers,” Feb 2023. [Online]. Available: https://doi.org/10.48550/arXiv.2302.11320
work page internal anchor Pith review Pith/arXiv arXiv doi:10.48550/arxiv.2302.11320 2023
-
[10]
Chemistry beyond the scale of exact diagonalization on a quantum-centric supercomputer,
J. Robledo-Moreno, M. Motta, H. Haas, A. Javadi-Abhari, P. Jurcevic, W. Kirby, S. Martiel, K. Sharma, S. Sharma, T. Shirakawa, I. Sitdikov, R.-Y. Sun, K. J. Sung, M. Takita, M. C. Tran, S. Yunoki, and A. Mezzacapo, “Chemistry beyond the scale of exact diagonalization on a quantum-centric supercomputer,”Science Advances, vol. 11, no. 25, p. eadu9991, Jun
-
[11]
Available: https://www.science.org/doi/abs/10.1126/sciadv.adu9991
[Online]. Available: https://www.science.org/doi/abs/10.1126/sciadv.adu9991
-
[12]
Successive approximations by the rayleigh-ritz variation method,
J. K. L. MacDonald, “Successive approximations by the rayleigh-ritz variation method,”Phys. Rev., vol. 43, pp. 830–833, May 1933. [Online]. Available: https://link.aps.org/doi/10.1103/PhysRev.43.830
-
[13]
J. Kenneth M. Merz, A. Shajan, D. Kaliakin, F. Liang, Y. Otsuka, T. Shirakawa, L. Broers, H. Xu, M. Tsuji, M. Sato, S. Yunoki, R. Wakizaka, Y. Kawashima, J. Doi, T. Itoko, H. Horii, T. Pellegrini, J. R. Moreno, K. J. Sung, E. Fejer, R. Walkup, S. Seelam, and M. Motta, “Crossing the 12,000-atom barrier with heterogeneous quantum-classical REFERENCES 29 sup...
work page internal anchor Pith review Pith/arXiv arXiv doi:10.48550/arxiv.2605.01138 2026
-
[14]
Linear-scaling quantum circuits for computational chemistry,
I. Magoulas and F. A. Evangelista, “Linear-scaling quantum circuits for computational chemistry,”Journal of Chemical Theory and Computation, vol. 19, no. 15, pp. 4815–4821, Aug 2023. [Online]. Available: https://doi.org/10.1021/acs.jctc.3c00376
-
[15]
Machine-learnedcompactsubspacegenerationforquantumselectedconfigurationinteraction within density matrix embedding framework,
A. K. Patra, K. S. V. Anurag, R. Maitra, R. Bhat, P. Sai Shankar, and G. Jaiganesh, “Machine-learnedcompactsubspacegenerationforquantumselectedconfigurationinteraction within density matrix embedding framework,” 2026, manuscript in preparation
2026
-
[16]
K. S. V. Anurag, A. K. Patra, M. Mukherjee, A. Shukla, P. Sai Shankar, R. Bhat, T. S. L. Radhika, and G. Jaiganesh, “Towards chemically accurate and scalable quantum simulations on iqm quantum hardware: A quantum-hpc hybrid approach,” Apr 2026. [Online]. Available: https://doi.org/10.48550/arXiv.2604.01983
-
[17]
M. Motta, K. J. Sung, K. B. Whaley, M. Head-Gordon, and J. Shee, “Bridging physical intuition and hardware efficiency for correlated electronic states: the local unitary cluster jastrow ansatz for electronic structure,”Chem. Sci., vol. 14, pp. 11213–11227, Sep 2023. [Online]. Available: http://dx.doi.org/10.1039/D3SC02516K
-
[18]
Density matrix embedding: A simple alternative to dynamical mean-field theory,
G. Knizia and G. K.-L. Chan, “Density matrix embedding: A simple alternative to dynamical mean-field theory,”Phys. Rev. Lett., vol. 109, p. 186404, Nov 2012. [Online]. Available: https://link.aps.org/doi/10.1103/PhysRevLett.109.186404
-
[19]
Structure of mpro from sars-cov-2 and discovery of its inhibitors,
Z. Jin, X. Du, Y. Xu, Y. Deng, M. Liu, Y. Zhao, B. Zhang, X. Li, L. Zhang, C. Peng, Y. Duan, J. Yu, L. Wang, K. Yang, F. Liu, R. Jiang, X. Yang, T. You, X. Liu, X. Yang, F. Bai, H. Liu, X. Liu, L. W. Guddat, W. Xu, G. Xiao, C. Qin, Z. Shi, H. Jiang, Z. Rao, and H. Yang, “Structure of mpro from sars-cov-2 and discovery of its inhibitors,”Nature, vol. 582, ...
-
[20]
Structural basis for the inhibition of sars-cov-2 main protease by antineoplastic drug carmofur,
Z. Jin, Y. Zhao, Y. Sun, B. Zhang, H. Wang, Y. Wu, Y. Zhu, C. Zhu, T. Hu, X. Du, Y. Duan, J. Yu, X. Yang, X. Yang, K. Yang, X. Liu, L. W. Guddat, G. Xiao, L. Zhang, H. Yang, and Z. Rao, “Structural basis for the inhibition of sars-cov-2 main protease by antineoplastic drug carmofur,”Nature Structural & Molecular Biology, vol. 27, no. 6, pp. 529–532, Jun. ...
-
[21]
T. Nisar, H. Sutherland-Foggio, and W. Husar, “Antiviral amantadine,”The Lancet Neurology, vol. 18, no. 12, p. 1080, Dec 2019. [Online]. Available: https://doi.org/10.1016/S1474-4422(19)30361-8
-
[22]
Cnot-efficient circuits for arbitrary rank many-body fermionic and qubit excitations,
I. Magoulas and F. A. Evangelista, “Cnot-efficient circuits for arbitrary rank many-body fermionic and qubit excitations,”Journal of Chemical Theory and Computation, vol. 19, no. 3, pp. 822–836, 2023. [Online]. Available: https://doi.org/10.1021/acs.jctc.2c01016
-
[23]
Über das paulische äquivalenzverbot,
P. Jordan and E. Wigner, “Über das paulische äquivalenzverbot,”Zeitschrift für Physik, vol. 47, no. 9, pp. 631–651, 1928. [Online]. Available: https://doi.org/10.1007/BF01331938
-
[24]
K. S. V. Anurag, A. K. Patra, V. D. Ghevade, P. Sai Shankar, R. Bhat, V. Raghavendra, R. Maitra, and G. Jaiganesh, “Resource estimation for vqe on small molecules: Impact of fermion mappings and hamiltonian reductions,”Journal of Computational Chemistry, vol. 47, no. 11, p. e70379, Apr 2026, e70379 3014834. [Online]. Available: https://onlinelibrary.wiley...
-
[25]
P. K. Barkoutsos, J. F. Gonthier, I. Sokolov, N. Moll, G. Salis, A. Fuhrer, M. Ganzhorn, D. J. Egger, M. Troyer, A. Mezzacapo, S. Filipp, and I. Tavernelli, “Quantum algorithms for electronic structure calculations: Particle-hole hamiltonian and optimized wave-function expansions,”Phys. Rev. A, vol. 98, p. 022322, Aug 2018. [Online]. Available: https://li...
-
[26]
Parallel implementation of high-fidelity multiqubit gates with neutral atoms,
H. Levine, A. Keesling, G. Semeghini, A. Omran, T. T. Wang, S. Ebadi, H. Bernien, M. Greiner, V. Vuletić, H. Pichler, and M. D. Lukin, “Parallel implementation of high-fidelity multiqubit gates with neutral atoms,”Phys. Rev. Lett., vol. 123, p. 170503, Oct
-
[27]
Available: https://link.aps.org/doi/10.1103/PhysRevLett.123.170503
[Online]. Available: https://link.aps.org/doi/10.1103/PhysRevLett.123.170503
-
[28]
High-fidelity parallel entangling gates on a neutral-atom quantum computer,
S. J. Evered, D. Bluvstein, M. Kalinowski, S. Ebadi, T. Manovitz, H. Zhou, S. H. Li, A. A. Geim, T. T. Wang, N. Maskara, H. Levine, G. Semeghini, M. Greiner, V. Vuletić, and M. D. Lukin, “High-fidelity parallel entangling gates on a neutral-atom quantum computer,”Nature, vol. 622, no. 7982, pp. 268–272, Oct 2023. [Online]. Available: https://doi.org/10.10...
-
[29]
Quantum 4:327, ISSN 2521-327X, ://dx.doi.org/10.22331/q-2020-09-21-327
L. Henriet, L. Beguin, A. Signoles, T. Lahaye, A. Browaeys, G.-O. Reymond, and C. Jurczak, “Quantum computing with neutral atoms,”Quantum, vol. 4, p. 327, sep 2020. [Online]. Available: https://doi.org/10.22331/q-2020-09-21-327
-
[30]
Benchmarking a machine-learning differential equations solver on a neutral-atom logical processor
P. Mathiot, E. Garnaoui, A.-U. Leriche, E. Philip, B. Albrecht, C. Briosne-Fréjaville, L. Cardarelli, A. Cornillot, G. Cournez, L. Couturier, J. D. Hond, R. E. Koussaifi, T. Eritzpokoff, F. Fasola, A. A. Gentile, C. Gyurik, C. Hamot, L. Henriet, G. Hercé, M. Kaicher, L. Lassablière, F.-M. L. Régent, E. Leroux, Y. Machu, H. Mamann, L. Ortiz, A. Paine, T. P...
work page internal anchor Pith review Pith/arXiv arXiv doi:10.48550/arxiv.2605.21276 2026
-
[31]
Parametrized multiqubit gates for neutral-atom quantum platforms,
M. Mohan, J. de Hond, and S. Kokkelmans, “Parametrized multiqubit gates for neutral-atom quantum platforms,”Phys. Rev. Appl., vol. 23, p. 054074, May 2025. [Online]. Available: https://link.aps.org/doi/10.1103/PhysRevApplied.23.054074
-
[32]
A. K. Patra, K. S. V. Anurag, P. Sai Shankar, R. Bhat, V. Raghavendra, R. Maitra, and G. Jaiganesh, “Quantum simulation of ligand-like molecules through sample-based quantum diagonalization in density matrix embedding framework,” Apr 2026. [Online]. Available: https://doi.org/10.48550/arXiv.2511.22158
work page internal anchor Pith review Pith/arXiv arXiv doi:10.48550/arxiv.2511.22158 2026
-
[33]
Unsupervised learning of distributions on binary vectors using two layer networks,
Y. Freund and D. Haussler, “Unsupervised learning of distributions on binary vectors using two layer networks,” inAdvances in Neural Information Processing Systems, J. Moody, S. Hanson, and R. Lippmann, Eds., vol. 4. Morgan-Kaufmann,
-
[34]
Available: https://proceedings.neurips.cc/paper_files/paper/1991/file/ 33e8075e9970de0cfea955afd4644bb2-Paper.pdf
[Online]. Available: https://proceedings.neurips.cc/paper_files/paper/1991/file/ 33e8075e9970de0cfea955afd4644bb2-Paper.pdf
1991
-
[35]
Restricted boltzmann machine learning for solving strongly correlated quantum systems,
Y. Nomura, A. S. Darmawan, Y. Yamaji, and M. Imada, “Restricted boltzmann machine learning for solving strongly correlated quantum systems,”Phys. Rev. B, vol. 96, p. 205152, Nov 2017. [Online]. Available: https://link.aps.org/doi/10.1103/PhysRevB.96.205152
-
[36]
Restricted boltzmann machines in quantum physics,
R. G. Melko, G. Carleo, J. Carrasquilla, and J. I. Cirac, “Restricted boltzmann machines in quantum physics,”Nature Physics, vol. 15, no. 9, pp. 887–892, 2019. [Online]. Available: https://doi.org/10.1038/s41567-019-0545-1
-
[37]
Solving the schrödinger equation in the configuration space with generative machine learning,
B. Herzog, B. Casier, S. Lebègue, and D. Rocca, “Solving the schrödinger equation in the configuration space with generative machine learning,”Journal of Chemical REFERENCES 31 Theory and Computation, vol. 19, no. 9, pp. 2484–2490, 2023. [Online]. Available: https://doi.org/10.1021/acs.jctc.2c01216
-
[38]
Physics- informed generative machine learning for accelerated quantum-centric supercomputing,
C. Patra, D. Mondal, S. Halder, D. Halder, M. R. Laskar, R. Goel, and R. Maitra, “Physics- informed generative machine learning for accelerated quantum-centric supercomputing,”
-
[39]
Available: https://doi.org/10.48550/arXiv.2512.06858
[Online]. Available: https://doi.org/10.48550/arXiv.2512.06858
-
[40]
A practical guide to density matrix embedding theory in quantum chemistry,
S. Wouters, C. A. Jiménez-Hoyos, Q. Sun, and G. K.-L. Chan, “A practical guide to density matrix embedding theory in quantum chemistry,”Journal of Chemical Theory and Computation, vol. 12, no. 6, pp. 2706–2719, Jun 2016. [Online]. Available: https://doi.org/10.1021/acs.jctc.6b00316
-
[41]
A. Shajan, D. Kaliakin, A. Mitra, J. Robledo Moreno, Z. Li, M. Motta, C. Johnson, A. A. Saki, S. Das, I. Sitdikov, A. Mezzacapo, and K. M. Merz, “Toward quantum-centric simulations of extended molecules: Sample-based quantum diagonalization enhanced with density matrix embedding theory,”Journal of Chemical Theory and Computation, vol. 21, no. 14, pp. 6801...
-
[42]
G. K.-L. Chan, M. Kállay, and J. Gauss, “State-of-the-art density matrix renormalization group and coupled cluster theory studies of the nitrogen binding curve,”The Journal of Chemical Physics, vol. 121, no. 13, pp. 6110–6116, Oct 2004. [Online]. Available: https://doi.org/10.1063/1.1783212
-
[43]
S. Barison, J. Robledo Moreno, and M. Motta, “Quantum-centric computation of molecular excited states with extended sample-based quantum diagonalization,”Quantum Science and Technology, vol. 10, no. 2, p. 025034, Feb 2025. [Online]. Available: https://doi.org/10.1088/2058-9565/adb781
-
[44]
Qulacs: a fast and versatile quantum circuit simulator for research purpose,
Y. Suzuki, Y. Kawase, Y. Masumura, Y. Hiraga, M. Nakadai, J. Chen, K. M. Nakanishi, K. Mitarai, R. Imai, S. Tamiya, T. Yamamoto, T. Yan, T. Kawakubo, Y. O. Nakagawa, Y. Ibe, Y. Zhang, H. Yamashita, H. Yoshimura, A. Hayashi, and K. Fujii, “Qulacs: a fast and versatile quantum circuit simulator for research purpose,”Quantum, vol. 5, p. 559, Oct
-
[45]
Available: https://doi.org/10.22331/q-2021-10-06-559
[Online]. Available: https://doi.org/10.22331/q-2021-10-06-559
-
[46]
E. R. Davidson, “The iterative calculation of a few of the lowest eigenvalues and corresponding eigenvectors of large real-symmetric matrices,”Journal of Computational Physics, vol. 17, no. 1, pp. 87–94, Jan 1975. [Online]. Available: https://doi.org/10.1016/0021-9991(75)90065-0 REFERENCES 32 Supplementary Information A. Molecular Geometries and Fragmenta...
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.