Enhancing quantum-classical configuration interaction methods using a neural-network classifier
Pith reviewed 2026-06-25 22:05 UTC · model grok-4.3
The pith
A neural-network classifier selects important determinants in configuration interaction calculations, matching traditional accuracy at lower computational cost.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
A neural-network classifier trained on labels from temporary diagonalizations within an active-learning loop can guide selection of important determinants in iterative configuration interaction, maintaining accuracy while reducing computational demands for both classical and hybrid quantum-classical variants.
What carries the argument
The neural-network classifier integrated into the iterative CI workflow through an active-learning loop that labels a random subset of candidates via temporary diagonalization at each step.
If this is right
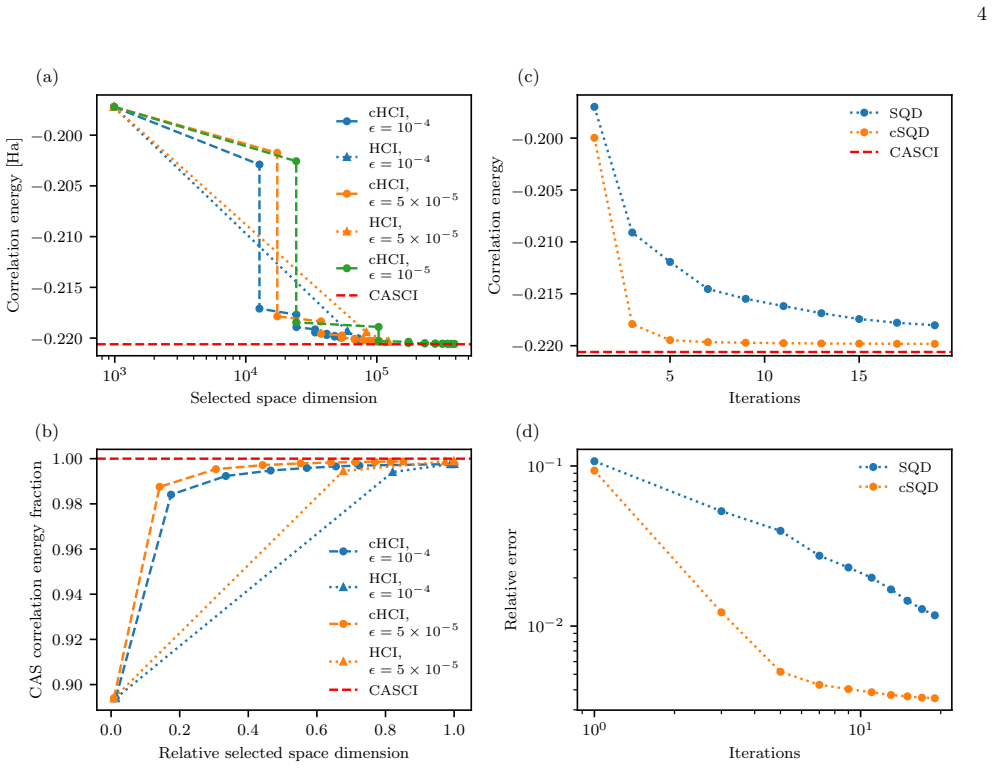
- Result parity is achieved with roughly a 5x reduction in memory and per-iteration cost for the classical cHCI variant.
- Convergence occurs in markedly fewer iterations for the quantum-classical cSQD variant.
- The framework is method-agnostic and can compress variational spaces for both classical and quantum CI algorithms.
- Classifier-assisted determinant selection acts as a lightweight tool for accelerating configuration interaction calculations.
Where Pith is reading between the lines
- The same active-learning loop could be applied to other variational quantum chemistry methods that rely on iterative determinant or configuration selection.
- If the classifier scales reliably, the approach might enable selected CI on systems where full enumeration of candidates is currently prohibitive.
- Combining the classifier with existing heuristics could further reduce the size of the labeled subset needed at each iteration.
Load-bearing premise
Labeling a random subset of candidate determinants via temporary diagonalisation at each iteration supplies training data sufficient for the neural-network classifier to generalize accurately to the full pool of remaining configurations without introducing systematic bias.
What would settle it
Running the method on a larger molecule or different basis set and finding that the classifier-selected variational space produces energies significantly above those from traditional selection or requires comparable or greater resources to reach the same accuracy.
Figures

read the original abstract
Selected configuration interaction methods achieve near-exact electronic structure calculations by iteratively constructing compact variational spaces, but their efficiency depends critically on the heuristics used to identify important determinants. Here, we introduce a data-driven selection framework that recasts determinant importance as a binary classification task and integrates a neural-network classifier into the iterative CI workflow through an active-learning loop. At each iteration, a random subset of candidate determinants is labelled via temporary diagonalisation, and the trained classifier guides selection of the remaining configurations. We demonstrate the utility of this framework for both classical and quantum CI methods by calculating the ground-state energy of a diatomic molecule. Our method achieves result parity with traditional configuration interaction methods at substantially lower computational cost: roughly a $\times 5$ reduction in memory and per-iteration cost for the classical cHCI variant, and convergence in markedly fewer iterations for the quantum-classical cSQD variant. These results establish classifier-assisted determinant selection as a lightweight, method-agnostic tool for compressing variational spaces and accelerating both classical and hybrid quantum-classical configuration interaction algorithms.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The paper introduces a data-driven framework for selected configuration interaction (CI) that recasts determinant importance as a binary classification task. A neural-network classifier is integrated into the iterative CI workflow via an active-learning loop: at each iteration a random subset of candidate determinants is labeled by temporary diagonalization, the classifier is trained on this subset, and it then guides selection of the remaining configurations. The method is demonstrated on both the classical cHCI variant and the quantum-classical cSQD variant for the ground-state energy of a diatomic molecule, with claims of result parity to traditional CI at substantially lower cost (roughly ×5 reduction in memory and per-iteration cost for cHCI; convergence in markedly fewer iterations for cSQD).
Significance. If the central performance claims hold, the work supplies a lightweight, method-agnostic tool for compressing variational spaces in both classical and hybrid quantum-classical CI algorithms. The active-learning integration of a neural-network classifier for determinant selection is a concrete, reproducible idea that could reduce reliance on hand-crafted heuristics while preserving variational accuracy.
major comments (2)
- [Abstract] Abstract: the headline claim of 'result parity with traditional configuration interaction methods at substantially lower computational cost' (including the specific ×5 memory/iteration reduction for cHCI and 'markedly fewer iterations' for cSQD) is stated without error bars, the identity of the diatomic molecule, convergence thresholds, or any ablation on classifier accuracy; these omissions make the quantitative claims impossible to verify from the supplied information.
- [Methods (active-learning loop)] Active-learning loop description (Methods): labeling only a random subset via temporary diagonalization supplies the training data for the neural-network classifier; no analysis is given on whether this subset is representative of the distribution of high-weight determinants or on the risk of systematic selection bias when the classifier is applied to the full remaining pool. This assumption is load-bearing for both the parity claim and the reported cost reductions.
minor comments (1)
- [Abstract] Abstract: the phrase 'a diatomic molecule' should be replaced by the specific chemical species and basis set used.
Simulated Author's Rebuttal
We thank the referee for their constructive comments on our manuscript. We address each major comment below, indicating planned revisions where appropriate.
read point-by-point responses
-
Referee: [Abstract] Abstract: the headline claim of 'result parity with traditional configuration interaction methods at substantially lower computational cost' (including the specific ×5 memory/iteration reduction for cHCI and 'markedly fewer iterations' for cSQD) is stated without error bars, the identity of the diatomic molecule, convergence thresholds, or any ablation on classifier accuracy; these omissions make the quantitative claims impossible to verify from the supplied information.
Authors: The abstract is written as a concise summary of the key findings. The diatomic molecule (H2), convergence thresholds, error bars on reported energies, and classifier ablation studies are all detailed in the Methods, Results, and Supplementary Information sections. We agree the abstract would benefit from greater specificity and will revise it to name the molecule and reference the supporting quantitative details and analyses provided in the main text. revision: yes
-
Referee: [Methods (active-learning loop)] Active-learning loop description (Methods): labeling only a random subset via temporary diagonalization supplies the training data for the neural-network classifier; no analysis is given on whether this subset is representative of the distribution of high-weight determinants or on the risk of systematic selection bias when the classifier is applied to the full remaining pool. This assumption is load-bearing for both the parity claim and the reported cost reductions.
Authors: This is a valid point regarding the active-learning procedure. The manuscript demonstrates that the final variational energies match those of standard CI, which indirectly supports the effectiveness of the random subset for training. However, we acknowledge that an explicit analysis of subset representativeness and potential bias would strengthen the presentation. We will add a dedicated paragraph (with supporting statistics or a small figure) comparing the weight distribution of determinants in the labeled subset versus the full candidate pool. revision: yes
Circularity Check
No circularity: performance claims are empirical, not forced by construction
full rationale
The method trains an NN classifier on labels obtained from independent temporary diagonalization of random candidate subsets, then uses the classifier to guide selection of the remaining determinants in an active-learning loop. The reported parity in ground-state energy and the claimed ×5 memory/iteration reductions for cHCI (or fewer iterations for cSQD) are presented as empirical outcomes measured against traditional CI baselines on a diatomic molecule. No equation, procedure, or self-citation in the abstract or described workflow makes the final variational energy or cost metric equivalent to the classifier's fitted parameters or training labels by definition. The central assumption (that the random subset supplies unbiased training data) is a methodological risk but does not constitute a circular reduction of the result to its inputs.
Axiom & Free-Parameter Ledger
Reference graph
Works this paper leans on
-
[1]
Ivanic and K
J. Ivanic and K. Ruedenberg, Identification of deadwood in configuration spaces through general direct configu- ration interaction, Theoretical Chemistry Accounts106, 339 (2001)
2001
-
[2]
Huron, J
B. Huron, J. P. Malrieu, and P. Rancurel, Iterative per- turbation calculations of ground and excited state en- ergies from multiconfigurational zeroth-order wavefunc- tions, The Journal of Chemical Physics58, 5745 (1973)
1973
-
[3]
N. M. Tubman, J. Lee, T. Y. Takeshita, M. Head- Gordon, and K. B. Whaley, A deterministic alternative to the full configuration interaction quantum monte carlo method, The Journal of Chemical Physics145, 044112 (2016)
2016
-
[4]
A. A. Holmes, N. M. Tubman, and C. J. Umrigar, Heat- bath configuration interaction: An efficient selected con- figuration interaction algorithm inspired by heat-bath sampling, Journal of Chemical Theory and Computation 12, 3674 (2016)
2016
-
[5]
Sharma, A
S. Sharma, A. A. Holmes, G. Jeanmairet, A. Alavi, and C. J. Umrigar, Semistochastic heat-bath configura- tion interaction method: Selected configuration interac- tion with semistochastic perturbation theory, Journal of Chemical Theory and Computation13, 1595 (2017)
2017
-
[6]
J. Li, M. Otten, A. A. Holmes, S. Sharma, and C. J. Umrigar, Fast semistochastic heat-bath configuration in- teraction, The Journal of Chemical Physics149, 214110 (2018)
2018
-
[7]
Robledo-Moreno, M
J. Robledo-Moreno, M. Motta, H. Haas, A. Javadi- Abhari, P. Jurcevic, W. Kirby, S. Martiel, K. Sharma, S. Sharma, T. Shirakawa, I. Sitdikov, R.-Y. Sun, K. J. Sung, M. Takita, M. C. Tran, S. Yunoki, and A. Mezza- capo, Chemistry beyond the scale of exact diagonaliza- tion on a quantum-centric supercomputer, Science Ad- vances11, eadu9991 (2025)
2025
-
[8]
P. Reinholdt, K. M. Ziems, E. R. Kjellgren, S. Co- riani, S. P. A. Sauer, and J. Kongsted, Critical limitations in quantum-selected configuration inter- action methods, Journal of Chemical Theory and Computation21, 6811 (2025), pMID: 40586729, https://doi.org/10.1021/acs.jctc.5c00375
-
[9]
J. P. Coe, Machine learning configuration interaction, Journal of Chemical Theory and Computation14, 5739 (2018)
2018
-
[10]
J. P. Coe, Machine learning configuration interaction for ab initio potential energy curves, Journal of Chemical Theory and Computation15, 6179 (2019)
2019
-
[11]
Jeong, C
W. Jeong, C. A. Gaggioli, and L. Gagliardi, Active learn- ing configuration interaction for excited-state calcula- tions of polycyclic aromatic hydrocarbons, Journal of Chemical Theory and Computation17, 7518 (2021)
2021
-
[12]
S. D. Pineda Flores, Chembot: A machine learning ap- proach to selective configuration interaction, Journal of Chemical Theory and Computation17, 4028 (2021)
2021
-
[13]
J. J. Goings, H. Hu, C. Yang, and X. Li, Reinforcement learning configuration interaction, Journal of Chemical Theory and Computation17, 5482 (2021)
2021
-
[14]
Herzog, B
B. Herzog, B. Casier, S. Leb` egue, and D. Rocca, Solving the schr¨ odinger equation in the configuration space with generative machine learning, Journal of Chemical Theory and Computation19, 2484 (2023)
2023
-
[15]
Bilous, A
P. Bilous, A. P´ alffy, and F. Marquardt, Deep-learning ap- proach for the atomic configuration interaction problem on large basis sets, Physical Review Letters131, 133002 (2023)
2023
-
[16]
Bilous, C
P. Bilous, C. Cheung, and M. Safronova, Neural-network approach to running high-precision atomic computations, Physical Review A110, 042818 (2024)
2024
-
[17]
Bilous, L
P. Bilous, L. Thirion, H. Menke, M. W. Haverkort, A. P´ alffy, and P. Hansmann, Neural-network-supported basis optimizer for the configuration interaction problem in quantum many-body clusters: Feasibility study and numerical proof, Physical Review B111, 035124 (2025)
2025
-
[18]
Y. L. A. Schmerwitz, L. Thirion, G. Levi, E. O. J´ onsson, P. Bilous, H. J´ onsson, and P. Hansmann, Neural-network-based selective configuration interaction approach to molecular electronic structure, Journal of Chemical Theory and Computation21, 2301 (2025)
2025
-
[19]
Q. Sun, T. C. Berkelbach, N. S. Blunt, G. H. Booth, S. Guo, Z. Li, J. Liu, J. D. McClain, E. R. Sayfut- yarova, S. Sharma, S. Wouters, and G. K.-L. Chan, PySCF: the python-based simulations of chemistry framework, WIREs Computational Molecular Science8, e1340 (2018)
2018
-
[20]
A. A. Saki, S. Barison, B. Fuller, J. R. Garrison, J. R. Glick, C. Johnson, A. Mezzacapo, J. Robledo-Moreno, M. Rossmannek, P. Schweigert, I. Sitdikov, and K. J. Sung, Qiskit addon: sample-based quantum diagonaliza- tion (2024)
2024
-
[21]
In a separate study [22], we show that, with currently available circuits and sampling techniques, we do not ob- serve advantages from quantum-hardware sampling rela- tive to this classical baseline
-
[22]
Varutti, S
G. Varutti, S. Zeni, J. Nespolo, and D. Trypogeorgos (2026), in preparation
2026
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.