Revealing the role of van der Waals interactions in thiophene adsorption on copper surfaces
Pith reviewed 2026-05-24 17:07 UTC · model grok-4.3
The pith
Non-local van der Waals forces control thiophene binding to copper surfaces as weak chemisorption.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
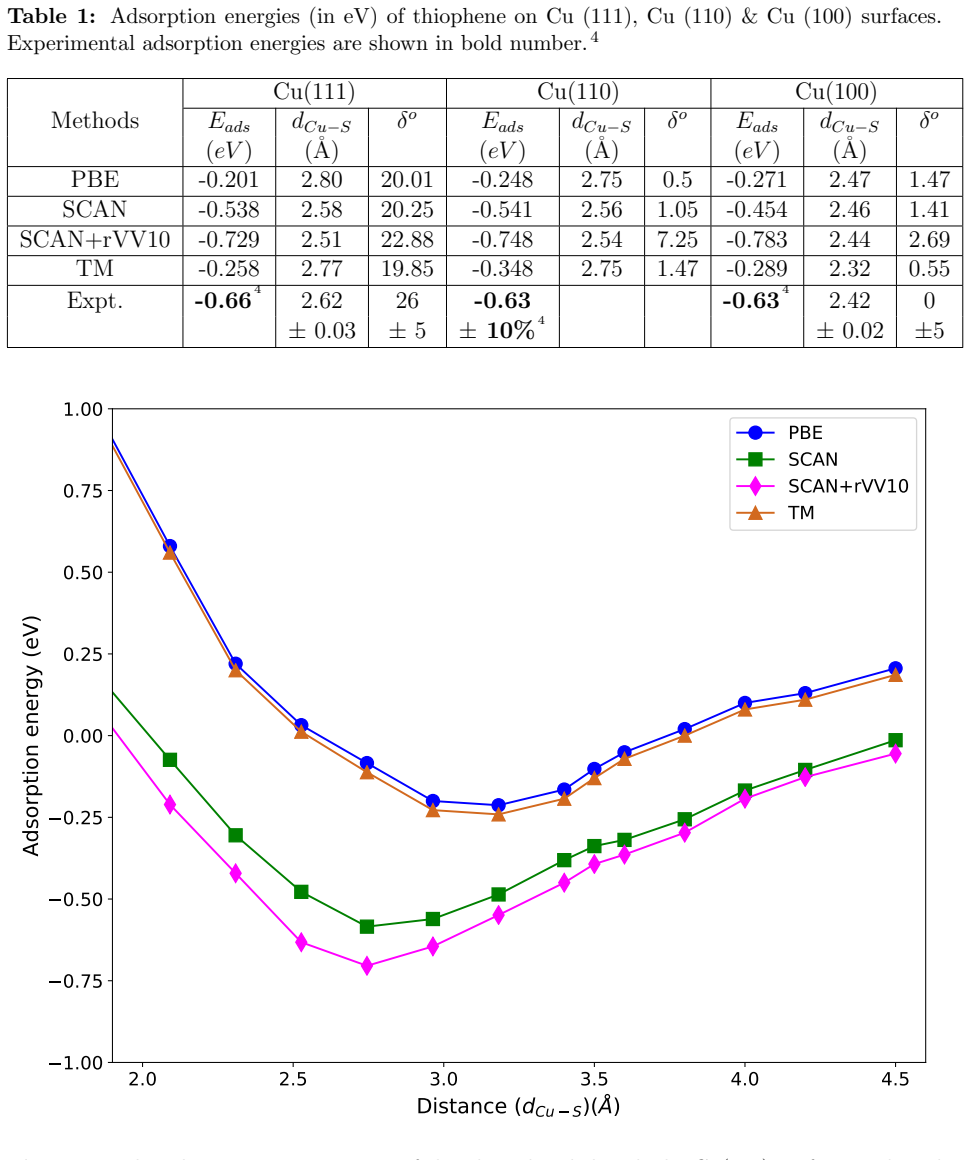
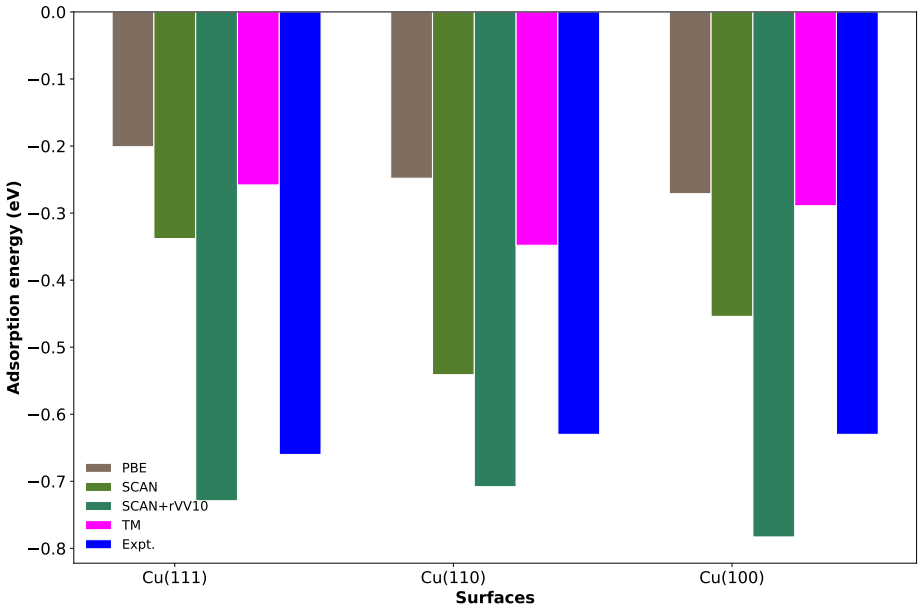
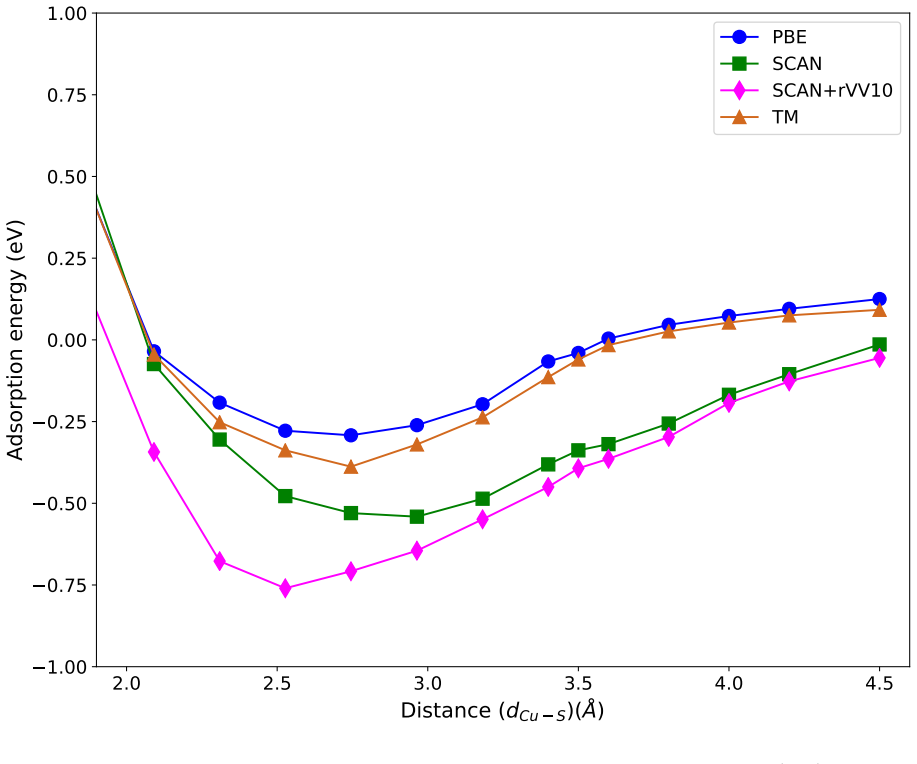
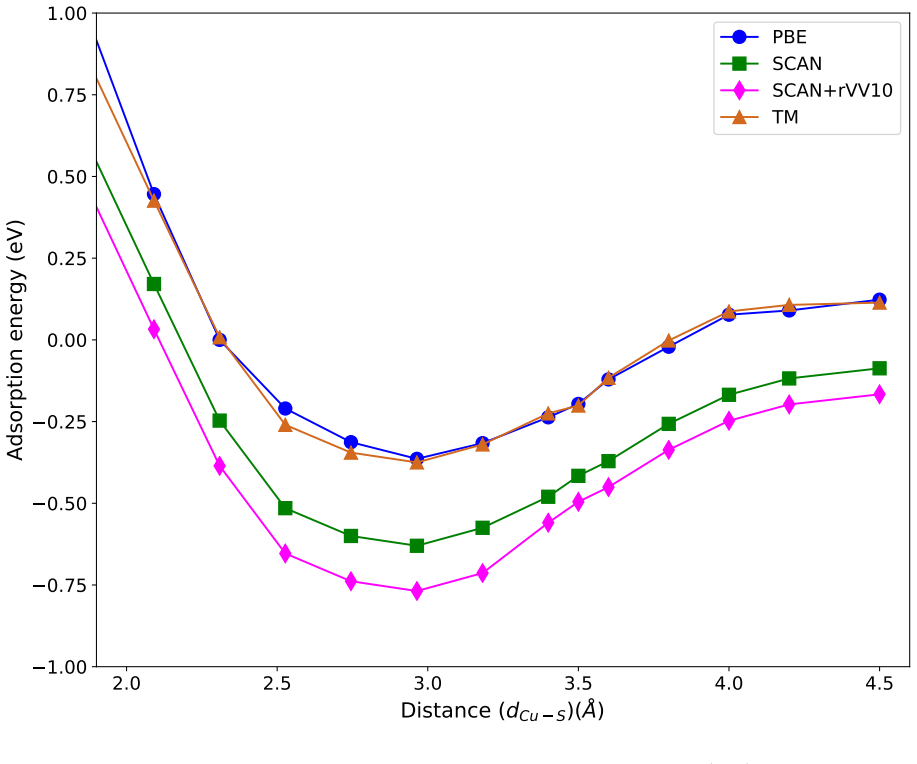
The weak chemisorption hypothesis of thiophene binding on the copper surface is well justified by the two meta-GGAs-based approximations, SCAN and SCAN+rVV10. Non-local dispersion interaction predominantly controls the bonding mechanism. Calculated adsorption energy curves show that this dispersion modifies the copper work function. Semi-local functionals without van der Waals correction misinterpret the process as physisorption, while SCAN benefits from fortuitous error cancellation and SCAN+rVV10 overestimates energies by an average of 16 percent due to excessive hybridization between sulfur p orbitals and copper d bands.
What carries the argument
Adsorption energy curves from the SCAN and SCAN+rVV10 meta-GGA functionals that incorporate non-local dispersion.
If this is right
- Semi-local functionals such as PBE-GGA and TM meta-GGA significantly underestimate adsorption energies and classify the interaction as physisorption.
- SCAN produces a numerically correct weak-chemisorption result through error cancellation rather than correct physics.
- SCAN+rVV10 overestimates adsorption energies by roughly 16 percent because the added dispersion term drives too much hybridization between sulfur p states and copper d bands.
- Inclusion of non-local dispersion alters the copper work function through the modified bonding.
Where Pith is reading between the lines
- Similar organic-molecule adsorption on other coinage-metal surfaces may require dispersion-inclusive functionals to avoid misclassifying the interaction type.
- Direct comparison of computed and measured work-function changes could test whether the over-hybridization seen in SCAN+rVV10 is physically realistic.
- The need for experimental benchmarks on this specific system suggests that other molecule-metal interfaces without such data may also suffer from unrecognized error cancellation.
Load-bearing premise
The chosen geometry with the sulfur atom directly above a copper atom is representative of the binding, and the functionals' error cancellation or hybridization behaviors can be trusted without experimental adsorption energy benchmarks for this system.
What would settle it
A measured adsorption energy for thiophene on copper that lies well outside the range predicted by SCAN and SCAN+rVV10 would falsify the claim that non-local dispersion controls the bonding.
Figures


read the original abstract
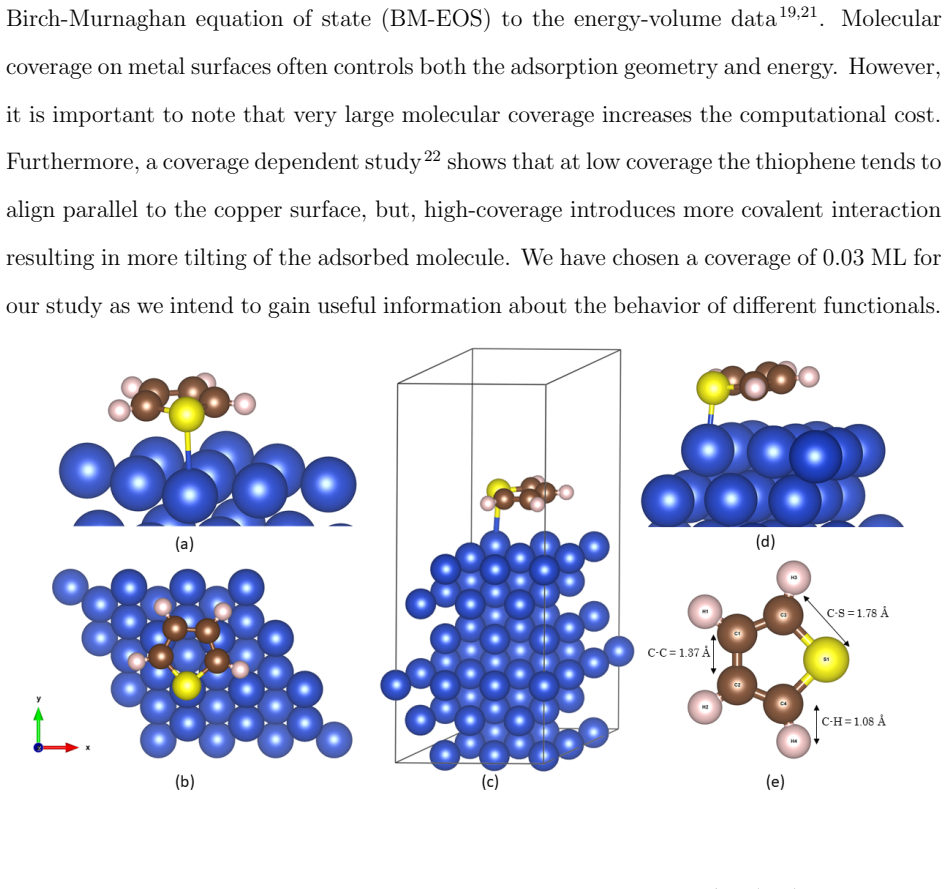
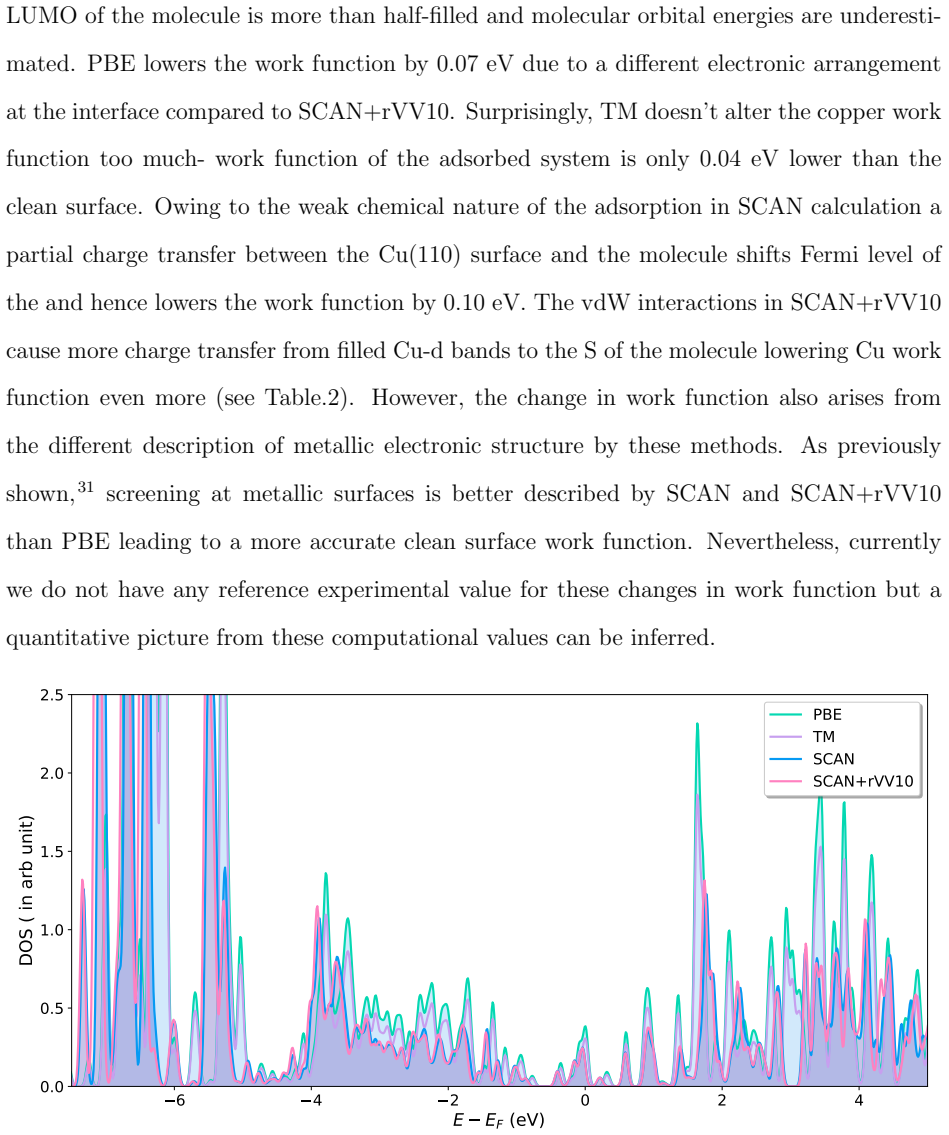
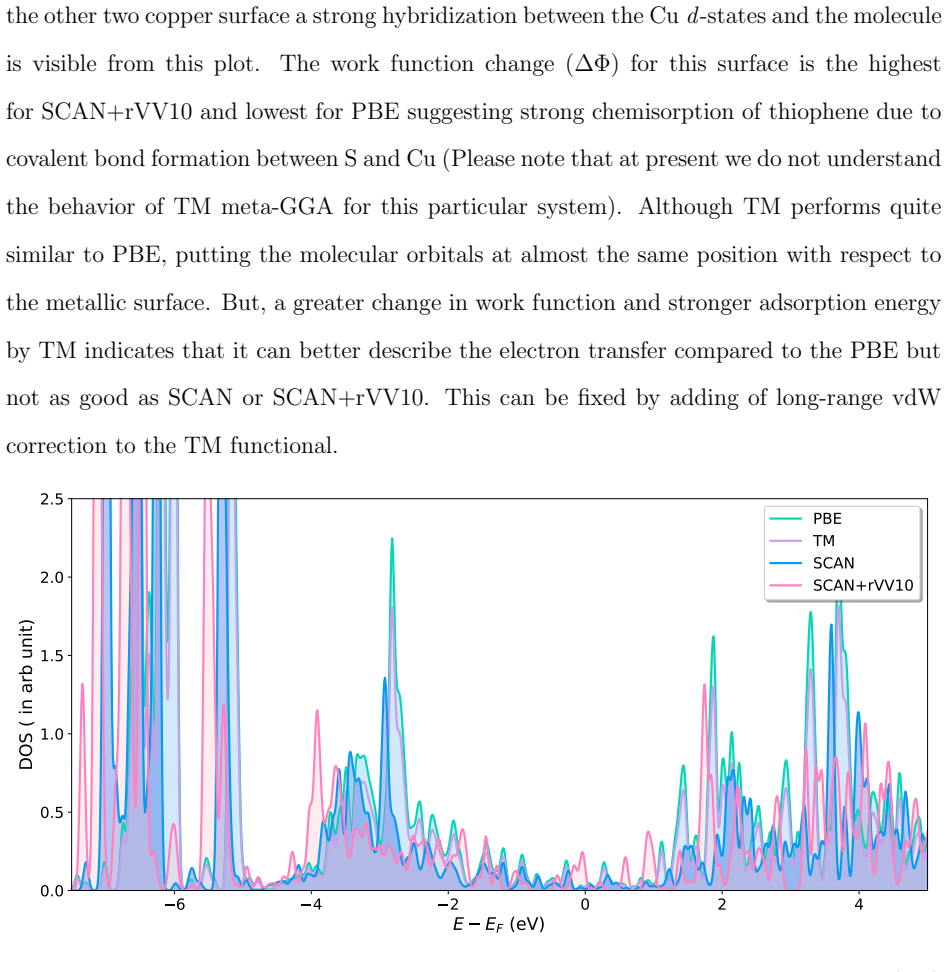
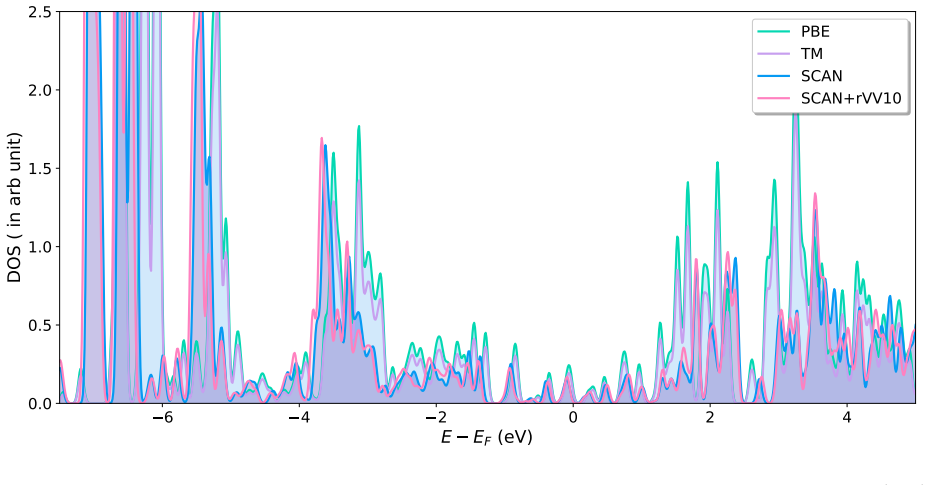
Accurate modeling of electronic and structural properties of organic molecule-metal interfaces are challenging problems because of the complicated electronic distribution of molecule and screening of charges at the metallic surface. This is also the reason why the organic/inorganic system can be engineered for several applications by fine-tuning the metallic work function. Here, we use density-functional theory (DFT) calculations with different level of functional approximations for a systematic study of thiophene interacting with Copper surfaces. In particular, we considered adsorbed structures with the thiophene molecule seated on the top site, with the S atom of the molecule located on the top of a Cu atom. In this work, we find that the weak chemisorption hypothesis of thiophene binding on the copper surface is well justified by the two meta-GGAs-based approximations, SCAN and SCAN+rVV10. PBE-GGA and TM meta-GGA describe it as a physisorption phenomenon by significantly underestimating the adsorption energies. Calculated adsorption energy curves reveal that non-local dispersion interaction between the molecule and metallic surface predominantly controls the bonding mechanism and thus, modifies the copper's work function. Our results imply that semi-local functionals without any kind of van der Waals (vdW) correction can often misinterpret this as physisorption, while, a fortuitous error cancellation can give a right description of this adsorption picture for a wrong reason as in the case of SCAN. The calculated density of states of the adsorbed molecule shows that the long-range vdW correction of SCAN+rVV10 causes more than enough hybridization between the \textit{p} orbitals of S atom and the copper \textit{d}-bands and therefore overestimates the adsorption energies by an average of 16\%.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript uses DFT with PBE, TM, SCAN, and SCAN+rVV10 functionals to examine thiophene adsorption on Cu surfaces in the S-on-top geometry. It concludes that SCAN and SCAN+rVV10 support a weak chemisorption picture dominated by non-local vdW interactions that also modify the work function, while PBE and TM incorrectly describe physisorption; SCAN+rVV10 is noted to overestimate energies by ~16% via excess p-d hybridization, and SCAN may succeed via error cancellation.
Significance. If the functional-dependent distinction between weak chemisorption and physisorption could be anchored to experiment, the work would usefully illustrate how semi-local functionals can misclassify bonding at organic-metal interfaces and highlight the role of dispersion. The computational trends across four functionals are internally consistent, but the central claim that non-local dispersion 'predominantly controls' the mechanism and that the meta-GGAs 'well justify' weak chemisorption remains unvalidated without measured adsorption energies or broader geometry sampling.
major comments (3)
- [Abstract / Results] Abstract and results discussion: the claim that SCAN and SCAN+rVV10 justify the weak chemisorption hypothesis rests on the functionals' outputs alone; the text itself states SCAN+rVV10 overestimates adsorption energies by an average of 16% and that SCAN may succeed only via 'fortuitous error cancellation,' yet no experimental adsorption energies for thiophene on Cu are cited to determine which description is physically correct.
- [Computational details / Results] Computational details / results: all analysis is restricted to the single S-on-top-Cu adsorption geometry; no checks are reported that this site is the global minimum or that other high-symmetry sites produce qualitatively different bonding mechanisms or vdW dominance.
- [Discussion] Discussion: the distinction between weak chemisorption (SCAN/SCAN+rVV10) and physisorption (PBE/TM) is presented as a functional-dependent outcome without external benchmarks, leaving the conclusion that 'non-local dispersion interaction ... predominantly controls the bonding mechanism' dependent on unvalidated error cancellation rather than falsifiable comparison to measured data.
minor comments (2)
- [Abstract] The abstract contains raw LaTeX commands (textit) that should be rendered in the final manuscript.
- [Results] No error bars, k-point or cutoff convergence data, or zero-point energy corrections are mentioned for the reported adsorption energies.
Simulated Author's Rebuttal
We thank the referee for the constructive comments on our comparative DFT study of thiophene adsorption on Cu surfaces. Our work examines how different functionals describe the bonding mechanism, with explicit acknowledgment of SCAN+rVV10 overestimation and possible error cancellation in SCAN. We address each major comment below, focusing on clarifications and targeted revisions to the language and discussion.
read point-by-point responses
-
Referee: [Abstract / Results] Abstract and results discussion: the claim that SCAN and SCAN+rVV10 justify the weak chemisorption hypothesis rests on the functionals' outputs alone; the text itself states SCAN+rVV10 overestimates adsorption energies by an average of 16% and that SCAN may succeed only via 'fortuitous error cancellation,' yet no experimental adsorption energies for thiophene on Cu are cited to determine which description is physically correct.
Authors: The study is a computational comparison of functionals to illustrate their differing treatments of dispersion and bonding at organic-metal interfaces. The manuscript already states the 16% overestimation by SCAN+rVV10 due to excess hybridization and notes possible error cancellation for SCAN. The 'weak chemisorption hypothesis is well justified' phrasing refers to the meta-GGAs' ability to capture non-local vdW forces leading to this picture, in contrast to PBE and TM. We will revise the abstract and results sections to emphasize that this is a functional-dependent outcome and that direct experimental adsorption energies would provide valuable external validation, without altering the core computational trends reported. revision: partial
-
Referee: [Computational details / Results] Computational details / results: all analysis is restricted to the single S-on-top-Cu adsorption geometry; no checks are reported that this site is the global minimum or that other high-symmetry sites produce qualitatively different bonding mechanisms or vdW dominance.
Authors: The S-on-top geometry was selected because it is the configuration reported in prior literature for thiophene on Cu surfaces. The analysis centers on the bonding mechanism and vdW role in this setup. We agree that confirming it as the global minimum or checking other sites would strengthen generality. We will add a clarifying statement in the computational details section noting this scope limitation and that vdW dominance is tied to the long-range interaction nature rather than site-specific effects. revision: partial
-
Referee: [Discussion] Discussion: the distinction between weak chemisorption (SCAN/SCAN+rVV10) and physisorption (PBE/TM) is presented as a functional-dependent outcome without external benchmarks, leaving the conclusion that 'non-local dispersion interaction ... predominantly controls the bonding mechanism' dependent on unvalidated error cancellation rather than falsifiable comparison to measured data.
Authors: The distinction is explicitly presented as arising from the functional approximations, with the text already discussing error cancellation and overestimation. The conclusion on non-local dispersion controlling the mechanism is drawn from the calculated energy curves and DOS showing hybridization changes with rVV10. We will revise the discussion to qualify the language, stressing the computational evidence for vdW importance while noting the absence of experimental benchmarks as a direction for future work. revision: partial
- Direct experimental adsorption energies or other measured data for validation, as the manuscript is a purely computational DFT study.
Circularity Check
No circularity; results are direct DFT outputs
full rationale
The paper reports adsorption energies, geometries, and DOS from standard DFT runs with PBE, TM, SCAN, and SCAN+rVV10 on a fixed S-on-top geometry. Conclusions follow directly from comparing these computed quantities across functionals; no parameters are fitted to the target adsorption energies, no self-referential definitions appear, and no load-bearing self-citations or imported uniqueness theorems are invoked. The derivation chain consists of external, reproducible electronic-structure calculations rather than any reduction to the paper's own inputs.
Axiom & Free-Parameter Ledger
axioms (1)
- domain assumption Density functional theory with the chosen functionals is sufficiently accurate to distinguish physisorption from weak chemisorption for this system.
Reference graph
Works this paper leans on
-
[1]
(1) Moth-Poulsen, K.; Bjørnholm, T. Nat. Nanotech. 2009, 4,
work page 2009
-
[2]
(2) Karth¨ auser, S.J. Phys. : Cond. Matt. 2011, 23, 013001. (3) Whitehurst, I. T., D. D.; Mochida, I. 1998, 42, 345 –
work page 2011
-
[3]
(4) Milligan, P.; MNamarra, J.; Murphy, B.; Cowie, B.; Lennon, D.; Kadodwala, M. Surf. Sci. 1998, 412-413,
work page 1998
-
[4]
(5) Rousseau, G.; Bovet, N.; Johnston, S.; Lennon, D.; Dhanak, V.; Kadodwala, M. Surf. Sci. 2002, 511,
work page 2002
-
[5]
(6) Orita, H.; Itoh, N. Surf. Sci. 2004, 550,
work page 2004
-
[6]
(7) Morin, C.; Eichler, A.; Hirschl, R.; Sautet, P.; Hafner, J. Surf. Sci. 2003, 540,
work page 2003
-
[7]
S.; Otero-de-la Roza, A.; Johnson, E
(8) Christian, M. S.; Otero-de-la Roza, A.; Johnson, E. R. J. Chem. Th. Comp. 2016, 12,
work page 2016
-
[8]
(9) Hu, L. H., Z-X.; Ji, W. Scientific Reports 2014, 4 . 18 (10) Lee, K.; Murray, E. D.; Kong, L.; Lundqvist, B.; Langreth, D. Phys. Rev. B 2010, 82, 081101. (11) Sony, P.; Puschnig, P.; Nabok, D.; Ambrosch-Draxl, C. Phys. Rev. Lett. 2007, 99, 176401. (12) Grimme, S. J. Comp. Chem. 2006, 27,
work page 2014
-
[9]
(13) Grimme, S.; Ehrlich, S.; Goerigk, L. J . Comp. Chem. 2011, 32,
work page 2011
-
[10]
(14) Goerigk, L.; Kruse, H.; Grimme, S. Chem. Phys. Chem. 2011, 12,
work page 2011
-
[11]
(15) Tonigold, K.; Grob, A. J. Chem. Phys. 2010, 132, 224701. (16) Callsen, M.; Atodiresei, N.; Caciuc, V.; Bl¨ ugel, S. Phys. Rev. B 2012, 86, 085439. (17) Perdew, J. P.; Schmidt, K. AIP Conference Proceedings 2001, 577, 1–20. (18) Sun, J.; Ruzsinszky, A.; Perdew, J. P. Phys. Rev. Lett. 2015, 115, 036402. (19) Tao, J.; Mo, Y. Phys. Rev. Lett. 2016, 117, ...
work page 2010
-
[12]
(24) Kresse, G.; Hafner, J. Phys. Rev. B 1994, 49, 14251. (25) Kresse, G.; Joubert, D. Phys. Rev. B 1999, 59,
work page 1994
-
[13]
(26) Bl¨ ochl, P. E.Phys. Rev. B 1994, 50, 17953. 19 (27) Hohenberg, P.; Kohn, W. Inhomogeneous Electron Gas. Phys Rev 1964, 136 . (28) Liu, W.; Ruiz, V. G.; Zhang, G.-X.; Santra, B.; Ren, X.; Scheffler, M.; Tkatchenko, A. New Journal of Physics 2013, 15, 053046. (29) Sun, J.; Remsing, R. C.; Zhang, Y.; Sun, Z.; Ruzsinszky, A.; Peng, H.; Yang, Z.; Paul, A.;...
-
[14]
(34) Tao, H., J.and Tang; Patra, A.; Bhattarai, P.; Perdew, J. P. Phys. Rev. B 2018, 97, 165403. (35) Tang, H.; Tao, J.; Ruzsinszky, A.; Perdew, J. P. J. Phys. Chem. C 2019, 123, 13748– 13757. 20
work page 2018
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.