Goodenough-Kanamori-Anderson rules in 2D magnet: A chemical trend in MCl2 with M=V, Mn, and Ni
Pith reviewed 2026-05-24 07:26 UTC · model grok-4.3
The pith
Monolayer NiCl2 is ferromagnetic while VCl2 and MnCl2 are antiferromagnetic, following Goodenough-Kanamori-Anderson rules.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
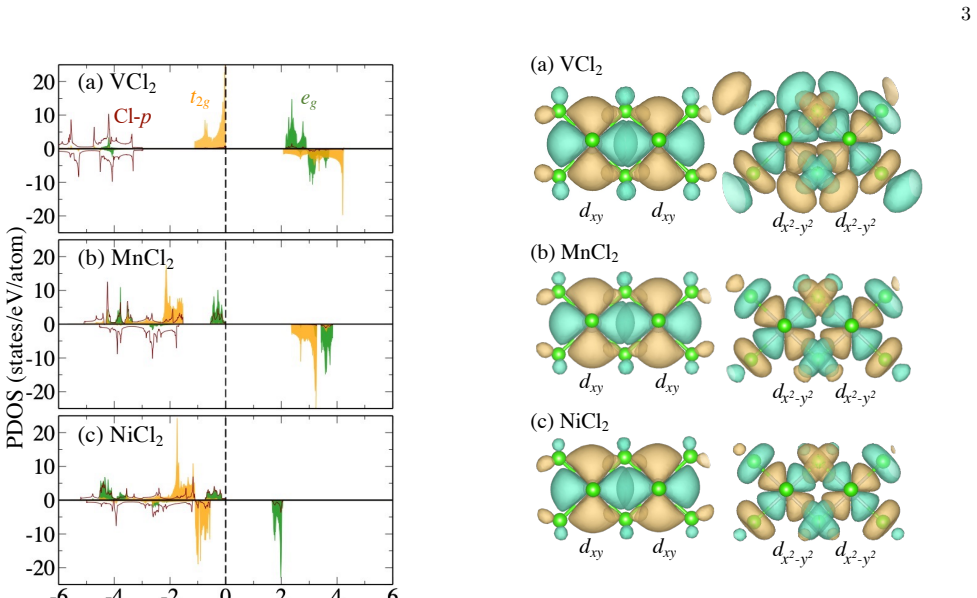
The magnetic stability in monolayer MCl2 (M=V, Mn, Ni) exhibits a distinct chemical trend: VCl2 and MnCl2 have antiferromagnetic ground states while NiCl2 has a ferromagnetic ground state. This ordering is explained by the Goodenough-Kanamori-Anderson rules together with the virtual-hopping process through hopping integrals between the 3d-orbital maximally localized Wannier functions, which highlights the competing roles of direct exchange and superexchange.
What carries the argument
Goodenough-Kanamori-Anderson rules applied to virtual-hopping integrals extracted from 3d-orbital maximally localized Wannier functions
If this is right
- Direct exchange dominates when orbitals are less than half-filled while superexchange dominates at half-filling in these triangular lattices.
- Chemical substitution of the transition-metal ion can switch the sign of the net exchange interaction in two-dimensional magnets.
- Interlayer coupling is not required for the Goodenough-Kanamori-Anderson mechanism to select the ground state.
- Maximally localized Wannier functions provide a practical route to quantify the hopping paths that set the exchange sign.
Where Pith is reading between the lines
- The same orbital-counting argument may predict magnetic order in other 2D metal halides once the d-electron filling is known.
- Device applications could exploit the switch from antiferromagnetic to ferromagnetic order by changing the metal species.
- The Wannier-based analysis could be repeated on strained or doped versions of the same monolayers to map how the trend evolves.
Load-bearing premise
Standard density-functional-theory calculations without Hubbard U or hybrid corrections reliably capture the relative energies of ferromagnetic versus antiferromagnetic states and that the extracted maximally localized Wannier functions accurately represent the dominant hopping paths.
What would settle it
Experimental measurement of the magnetic ground state in isolated monolayer samples of VCl2, MnCl2, or NiCl2, or recalculation of the same energy differences with hybrid functionals or added Hubbard corrections.
Figures


read the original abstract
Density-functional-theory calculations were performed to investigate the magnetism in a series of triangular-lattice monolayer MCl2 (M=V, Mn, and Ni). The magnetic stability manifests a distinct chemical trend; VCl2 and MnCl2 show the antiferromagnetic ground states and NiCl2 shows the ferromagnetic ground state. The microscopic mechanism behind the magnetic interaction is explained by the so-called Goodenough-Kanamori-Anderson rules and by the virtual-hopping process through the hopping integrals between the 3d-orbital maximally localized Wannier functions. Our result highlights the role of the direct exchange interaction and the superexchange interaction in the magnetic stabilization in two-dimensional magnets.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript reports density-functional theory calculations on the magnetic properties of triangular-lattice monolayer MCl2 (M = V, Mn, Ni). It claims a chemical trend with antiferromagnetic ground states for VCl2 and MnCl2 and a ferromagnetic ground state for NiCl2, interpreted through the Goodenough-Kanamori-Anderson rules and virtual-hopping processes extracted from 3d-orbital maximally localized Wannier functions. The abstract states that the results highlight the roles of direct exchange and superexchange in 2D magnetic stabilization.
Significance. If the DFT results hold, the work would establish a clear chemical trend in magnetic ordering for these 2D chlorides and provide a microscopic mechanism linking orbital hopping to the observed stability, which could inform design of related 2D magnets.
major comments (2)
- [Abstract] Abstract: The central claim of a distinct chemical trend in magnetic ground states rests on DFT total-energy comparisons, yet no numerical energy differences (e.g., E_FM - E_AFM), error bars, or convergence data are reported. This is load-bearing because the trend and subsequent GKA interpretation cannot be assessed without these values.
- [Computational methods] Computational methods (inferred from abstract and skeptic note): The exchange-correlation functional and any Hubbard U values are unspecified. For open-shell 3d systems, semilocal functionals commonly invert FM/AFM ordering due to self-interaction error; without U-sensitivity tests or hybrid-functional benchmarks, the reported ordering for VCl2/MnCl2/NiCl2 cannot be considered reliable.
minor comments (1)
- [Abstract] The abstract mentions 'maximally localized Wannier functions' but does not indicate how many bands were included or the quality of the Wannierization (e.g., spread values).
Simulated Author's Rebuttal
We thank the referee for the constructive comments, which help strengthen the manuscript. We address each major point below and have revised the manuscript accordingly to include the requested numerical data and methodological details.
read point-by-point responses
-
Referee: [Abstract] Abstract: The central claim of a distinct chemical trend in magnetic ground states rests on DFT total-energy comparisons, yet no numerical energy differences (e.g., E_FM - E_AFM), error bars, or convergence data are reported. This is load-bearing because the trend and subsequent GKA interpretation cannot be assessed without these values.
Authors: We agree that explicit numerical values are necessary to substantiate the claimed chemical trend. In the revised manuscript we have added Table I reporting the total-energy differences ΔE = E_AFM − E_FM (meV/f.u.) for each monolayer, together with the k-point mesh and plane-wave cutoff used, and a short convergence test showing that the sign of ΔE is stable to within 1 meV when the cutoff is increased by 20 %. These values confirm antiferromagnetic ordering for VCl2 and MnCl2 and ferromagnetic ordering for NiCl2, thereby supporting the subsequent GKA analysis. revision: yes
-
Referee: [Computational methods] Computational methods (inferred from abstract and skeptic note): The exchange-correlation functional and any Hubbard U values are unspecified. For open-shell 3d systems, semilocal functionals commonly invert FM/AFM ordering due to self-interaction error; without U-sensitivity tests or hybrid-functional benchmarks, the reported ordering for VCl2/MnCl2/NiCl2 cannot be considered reliable.
Authors: The original submission indeed omitted an explicit statement of the functional and U value in the main text. We have now inserted a dedicated “Computational Methods” paragraph stating that all calculations employed the PBE functional with an on-site U = 3 eV applied to the M 3d orbitals (U chosen from literature values for the corresponding bulk compounds). To address reliability concerns we have added a new subsection that (i) varies U from 0 to 5 eV and shows the magnetic ground-state ordering remains unchanged, and (ii) reports single-point PBE0 hybrid-functional calculations on 2×2 supercells that reproduce the same FM/AFM trend. These tests are now included in the revised manuscript. revision: yes
Circularity Check
No circularity: forward DFT computation with post-hoc interpretation via established external rules
full rationale
The paper reports standard DFT total-energy comparisons for FM vs AFM configurations in MCl2 monolayers, followed by MLWF extraction to obtain hopping integrals that are then interpreted using the classic Goodenough-Kanamori-Anderson rules. No equations define a target quantity in terms of itself, no parameters are fitted to the reported magnetic ordering or hoppings and then relabeled as predictions, and no load-bearing premise rests on a self-citation chain. The GKA rules predate the authors by decades and are invoked only for qualitative explanation after the computations. The derivation chain is therefore self-contained against external benchmarks (DFT energies and Wannier hoppings) and receives score 0.
Axiom & Free-Parameter Ledger
free parameters (1)
- DFT exchange-correlation functional and any Hubbard U values
axioms (1)
- domain assumption Density functional theory in its chosen form yields reliable relative energies between ferromagnetic and antiferromagnetic configurations in these 2D halides
Lean theorems connected to this paper
-
IndisputableMonolith/Cost/FunctionalEquation.leanwashburn_uniqueness_aczel unclear?
unclearRelation between the paper passage and the cited Recognition theorem.
The microscopic mechanism behind the magnetic interaction is explained by the so-called Goodenough-Kanamori-Anderson rules and by the virtual-hopping process through the hopping integrals between the 3d-orbital maximally localized Wannier functions.
-
IndisputableMonolith/Foundation/RealityFromDistinction.leanreality_from_one_distinction unclear?
unclearRelation between the paper passage and the cited Recognition theorem.
Density-functional-theory calculations were performed... Hubbard U potential, U = 1.8 eV...
What do these tags mean?
- matches
- The paper's claim is directly supported by a theorem in the formal canon.
- supports
- The theorem supports part of the paper's argument, but the paper may add assumptions or extra steps.
- extends
- The paper goes beyond the formal theorem; the theorem is a base layer rather than the whole result.
- uses
- The paper appears to rely on the theorem as machinery.
- contradicts
- The paper's claim conflicts with a theorem or certificate in the canon.
- unclear
- Pith found a possible connection, but the passage is too broad, indirect, or ambiguous to say the theorem truly supports the claim.
Reference graph
Works this paper leans on
-
[2]
R. B. Murray, Phys. Rev. 128, 1570 (1962)
work page 1962
- [3]
-
[4]
M. Gibertini, M. Koperski, A. F. Morpurgo, and K. S. Novoselov, Nat. Nanotechnol. 14, 408 (2019)
work page 2019
-
[5]
Q. H. Wang, A. Bedoya-Pinto, M. Blei, A. H. Dismukes, A. Hamo, S. Jenkins, M. Koperski, Y. Liu, Q.-C. Sun, E. J. Telford, H. H. Kim, M. Augustin, U. Vool, J.- X. Yin, L. H. Li, A. Falin, C. R. Dean, F. Casanova, R. F. L. Evans, M. Chshiev, A. Mishchenko, C. Petrovic, R. He, L. Zhao, A. W. Tsen, B. D. Gerardot, M. Brotons- Gisbert, Z. Guguchia, X. Roy, S. ...
work page 2022
-
[6]
M. A. McGuire, Crystals 7, 121 (2017)
work page 2017
-
[7]
C. Gong, L. Li, Z. Li, H. Ji, A. Stern, Y. Xia, T. Cao, W. Bao, C. Wang, Y. Wang, Z. Q. Qiu, R. J. Cava, S. G. Louie, J. Xia, and X. Zhang, Nature 546, 265 (2017)
work page 2017
- [8]
- [9]
-
[10]
S. Son, M. J. Coak, N. Lee, J. Kim, T. Y. Kim, H. Hami- dov, H. Cho, C. Liu, D. M. Jarvis, P. A. C. Brown, J. H. Kim, C.-H. Park, D. I. Khomskii, S. S. Saxena, and J.-G. Park, Phys. Rev. B 99, 041402(R) (2019)
work page 2019
-
[11]
T. P. T. Nguyen, K. Yamauchi, T. Oguchi, D. Amoroso, and S. Picozzi, Phys. Rev. B 104, 014414 (2021)
work page 2021
-
[12]
K. Kim, S. Y. Lim, J.-U. Lee, S. Lee, T. Y. Kim, K. Park, G. S. Jeon, C.-H. Park, J.-G. Park, and H. Cheong, Nat. Commun. 10, 345 (2019)
work page 2019
-
[13]
K. Kim, S. Y. Lim, J. Kim, J.-U. Lee, S. Lee, P. Kim, K. Park, S. Son, C.-H. Park, J.-G. Park, and H. Cheong, 2D Materials 6, 041001 (2019)
work page 2019
- [14]
- [15]
-
[16]
V. V. Kulish and W. Huang, J. Mater. Chem. C 5, 8734 (2017)
work page 2017
-
[17]
J. Luo, G. Xiang, Y. Tang, K. Ou, and X. Chen, J. Appl. Phys. 128, 113901 (2020)
work page 2020
- [18]
- [19]
- [20]
-
[21]
Q. Song, C. A. Occhialini, E. Erge¸ cen, B. Ilyas, D. Amoroso, P. Barone, J. Kapeghian, K. Watanabe, T. Taniguchi, A. S. Botana, S. Picozzi, N. Gedik, and R. Comin, Nature 602, 601 (2022)
work page 2022
-
[22]
A. O. Fumega and J. L. Lado, 2D Materials 9, 025010 (2022)
work page 2022
- [24]
- [25]
-
[26]
H. Kadowaki, K. Ubukoshi, K. Hirakawa, J. L. Mart´ ınez, and G. Shirane, J. Phys. Soc. Jpn. 56, 4027 (1987)
work page 1987
-
[27]
T. B. Prayitno and F. Ishii, J. Phys. Soc. Jpn. 88, 104705 (2019)
work page 2019
-
[28]
D. G. Wiesler, M. Suzuki, I. S. Suzuki, and N. Rosov, Phys. Rev. B 55, 6382 (1997)
work page 1997
- [29]
-
[30]
P. E. Bl¨ ochl, Phys. Rev. B50, 17953 (1994)
work page 1994
-
[31]
J. P. Perdew, K. Burke, and M. Ernzerhof, Phys. Rev. Lett. 77, 3865 (1996)
work page 1996
-
[32]
A. I. Liechtenstein, V. I. Anisimov, and J. Zaanen, Phys. Rev. B 52, R5467 (1995)
work page 1995
-
[33]
A. A. Mostofi, J. R. Yates, Y.-S. Lee, I. Souza, D. Van- derbilt, and N. Marzari, Comput. Phys. Commun. 178, 685 (2008)
work page 2008
-
[34]
N. Marzari, A. A. Mostofi, J. R. Yates, I. Souza, and D. Vanderbilt, Rev. Mod. Phys. 84, 1419 (2012)
work page 2012
-
[35]
G. Pizzi, V. Vitale, R. Arita, S. Bl¨ ugel, F. Freimuth, G. G´ eranton, M. Gibertini, D. Gresch, C. Johnson, T. Koretsune, J. Iba˜ nez-Azpiroz, H. Lee, J.-M. Lihm, D. Marchand, A. Marrazzo, Y. Mokrousov, J. I. Mustafa, Y. Nohara, Y. Nomura, L. Paulatto, S. Ponc´ e, T. Pon- weiser, J. Qiao, F. Th¨ ole, S. S. Tsirkin, M. Wierzbowska, N. Marzari, D. Vanderbi...
work page 2020
-
[36]
A. S. Botana and M. R. Norman, Phys. Rev. Materials 3, 044001 (2019)
work page 2019
-
[37]
R. H. Busey and W. F. Giauque, J. Am. Chem. Soc. 74, 4443 (1952)
work page 1952
- [38]
-
[39]
A. Liechtenstein, M. Katsnelson, V. Antropov, and V. Gubanov, J. Magn. Magn. Mater. 67, 65 (1987)
work page 1987
-
[40]
D. Okuyama, K. Yamauchi, H. Sakai, Y. Taguchi, Y. Tokura, K. Sugimoto, T. J. Sato, and T. Oguchi, Phys. Rev. Res. 2, 033038 (2020)
work page 2020
-
[41]
J. B. Goodenough, Phys. Rev. 100, 564 (1955)
work page 1955
- [42]
-
[43]
P. W. Anderson, Phys. Rev. 79, 350 (1950)
work page 1950
-
[44]
D. I. Khomskii, Transition Metal Compounds (Cam- bridge University Press, 2014)
work page 2014
-
[45]
M. W. Haverkort, M. Zwierzycki, and O. K. Andersen, Phys. Rev. B 85, 165113 (2012)
work page 2012
- [46]
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.