Representational Alignment with Chemical Induced Fit for Molecular Relational Learning
Pith reviewed 2026-05-25 08:25 UTC · model grok-4.3
The pith
ReAlignFit introduces chemical induced fit bias to align substructure representations and stabilize molecular relational learning on shifted data.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
ReAlignFit enhances the stability of molecular relational learning by dynamically aligning substructure representations through a chemical induced fit-based inductive bias; the bias is realized by a correction function that reconstructs substructure edges to simulate conformational changes, and the alignment is further refined by the subgraph information bottleneck that selects substructure pairs with high functional compatibility for generating molecular embeddings.
What carries the argument
Bias correction function based on substructure edge reconstruction, which supplies the chemical induced fit inductive bias that aligns representations between substructure pairs by simulating dynamic conformational changes.
If this is right
- ReAlignFit outperforms prior models on nine molecular relational learning datasets in two standard tasks.
- The method produces measurably higher stability when test distributions shift in functional-group rules or molecular scaffolds.
- The induced-fit alignment step can be added to existing graph encoders without changing their core architecture.
Where Pith is reading between the lines
- The same edge-reconstruction bias could be tested on non-molecular relational tasks that also involve conformational or structural adaptation.
- Replacing the subgraph bottleneck with other information-bottleneck variants would reveal whether the stability gain depends on the specific bottleneck formulation.
- Explicit modeling of induced fit may increase interpretability by making the learned substructure compatibilities traceable to chemical rules.
Load-bearing premise
The bias correction function based on substructure edge reconstruction accurately simulates chemical conformational changes and supplies a useful inductive bias that improves alignment without introducing artifacts.
What would settle it
Running ReAlignFit and an attention-only baseline on a held-out set of rule-shifted or scaffold-shifted molecular pairs and finding no gain in alignment consistency or downstream task stability would falsify the claim.
Figures





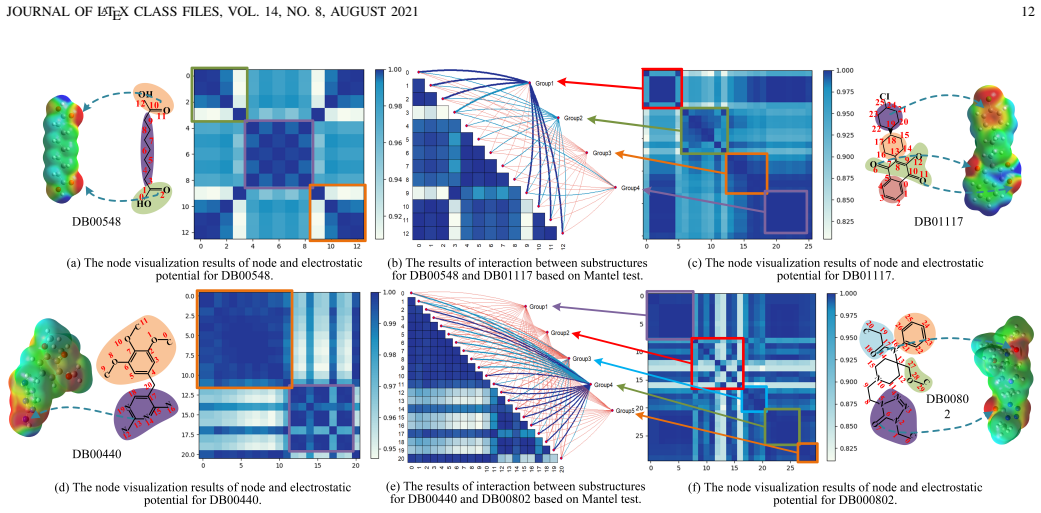

read the original abstract
Molecular Relational Learning (MRL) is widely applied in natural sciences to predict relationships between molecular pairs by extracting structural features. The representational similarity between substructure pairs determines the functional compatibility of molecular binding sites. Nevertheless, aligning substructure representations by attention mechanisms lacks guidance from chemical knowledge, resulting in unstable model performance in chemical space (\textit{e.g.}, functional group, scaffold) shifted data. With theoretical justification, we propose the \textbf{Re}presentational \textbf{Align}ment with Chemical Induced \textbf{Fit} (ReAlignFit) to enhance the stability of MRL. ReAlignFit dynamically aligns substructure representation in MRL by introducing chemical Induced Fit-based inductive bias. In the induction process, we design the Bias Correction Function based on substructure edge reconstruction to align representations between substructure pairs by simulating chemical conformational changes (dynamic combination of substructures). ReAlignFit further integrates the Subgraph Information Bottleneck during fit process to refine and optimize substructure pairs exhibiting high chemical functional compatibility, leveraging them to generate molecular embeddings. Experimental results on nine datasets demonstrate that ReAlignFit outperforms state-of-the-art models in two tasks and significantly enhances model's stability in both rule-shifted and scaffold-shifted data distributions.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The paper proposes ReAlignFit for molecular relational learning (MRL), which dynamically aligns substructure representations by introducing a chemical induced-fit inductive bias via a bias correction function based on substructure edge reconstruction (to simulate conformational changes), combined with a subgraph information bottleneck to refine high-compatibility pairs for molecular embeddings. It claims theoretical justification for this approach and reports outperformance over state-of-the-art models on nine datasets in two tasks, plus significantly improved stability under rule-shifted and scaffold-shifted distributions.
Significance. If the edge-reconstruction bias correction supplies a chemically faithful inductive bias rather than an unprincipled regularizer, the method could meaningfully address instability in MRL under distribution shifts common in molecular applications such as binding prediction. The multi-dataset evaluation is a positive empirical feature, though its value depends on controls for the shifts and statistical reporting.
major comments (2)
- [Induction process] Induction process description: the bias correction function is defined via substructure edge reconstruction and asserted to simulate chemical conformational changes for induced-fit alignment, yet no derivation, energy model, or reference to physical quantities (e.g., dihedral potentials or steric effects) is supplied to ground this correspondence; this assumption is load-bearing for the central claim of a 'chemical Induced Fit-based inductive bias' and the subsequent stability gains.
- [Fit process] Subgraph Information Bottleneck integration: the paper states that this step refines pairs with high chemical functional compatibility, but the interaction between the bottleneck objective and the preceding bias-correction term is not shown to preserve the claimed chemical grounding or to avoid introducing artifacts under the distribution shifts tested.
minor comments (2)
- The abstract and methods would benefit from explicit citation of prior induced-fit modeling literature to contextualize the proposed simulation.
- [Experimental results] Experimental reporting should include error bars, number of runs, and precise definitions of the rule-shifted and scaffold-shifted splits to allow reproducibility assessment.
Simulated Author's Rebuttal
We thank the referee for the constructive feedback on the chemical grounding of our approach. We address each major comment below.
read point-by-point responses
-
Referee: [Induction process] Induction process description: the bias correction function is defined via substructure edge reconstruction and asserted to simulate chemical conformational changes for induced-fit alignment, yet no derivation, energy model, or reference to physical quantities (e.g., dihedral potentials or steric effects) is supplied to ground this correspondence; this assumption is load-bearing for the central claim of a 'chemical Induced Fit-based inductive bias' and the subsequent stability gains.
Authors: We agree that the manuscript would benefit from greater clarity on this point. The bias correction is presented as an inductive bias inspired by the induced-fit concept rather than a direct physical simulation derived from energy models. The edge reconstruction acts as a proxy to encourage dynamic substructure alignment. The theoretical justification in the paper centers on improved stability under shifts, which is validated empirically. We will revise the relevant sections to explicitly state the modeling assumptions, add citations to induced-fit literature, and avoid any implication of quantitative physical derivation. revision: yes
-
Referee: [Fit process] Subgraph Information Bottleneck integration: the paper states that this step refines pairs with high chemical functional compatibility, but the interaction between the bottleneck objective and the preceding bias-correction term is not shown to preserve the claimed chemical grounding or to avoid introducing artifacts under the distribution shifts tested.
Authors: The bottleneck is applied to pairs already aligned via the bias correction to emphasize high-compatibility substructures. We acknowledge that the manuscript does not include an explicit analysis of their interaction or potential artifacts. We will revise by adding an analysis (e.g., via ablations or compatibility metrics) on the rule- and scaffold-shifted datasets to demonstrate that the combined objective preserves the alignment benefits. revision: yes
Circularity Check
No circularity: derivation chain not inspectable from abstract; no equations or self-referential reductions shown
full rationale
The abstract asserts 'theoretical justification' and describes a Bias Correction Function based on substructure edge reconstruction that 'simulates chemical conformational changes', but provides no equations, derivation steps, or citations that could be checked for self-definition, fitted-input prediction, or self-citation load-bearing. No load-bearing step reduces by construction to its inputs. The central inductive bias is presented as an external chemical motivation rather than derived from the model's own outputs or prior self-citations. This is the expected honest non-finding when the text supplies no derivation chain to walk.
Axiom & Free-Parameter Ledger
axioms (1)
- domain assumption Chemical induced fit can be simulated by substructure edge reconstruction to align representations.
Lean theorems connected to this paper
-
IndisputableMonolith/Cost/FunctionalEquation.leanwashburn_uniqueness_aczel unclear?
unclearRelation between the paper passage and the cited Recognition theorem.
we design the Bias Correction Function based on substructure edge reconstruction to align representations between substructure pairs by simulating chemical conformational changes
-
IndisputableMonolith/Foundation/RealityFromDistinction.leanreality_from_one_distinction unclear?
unclearRelation between the paper passage and the cited Recognition theorem.
Theorem II.1 ... P(Gx, Gy; Y) ≥ P(Gc; Y|Gn) ... ε measures the similarity between true probability, learning probability, and confounding probability
What do these tags mean?
- matches
- The paper's claim is directly supported by a theorem in the formal canon.
- supports
- The theorem supports part of the paper's argument, but the paper may add assumptions or extra steps.
- extends
- The paper goes beyond the formal theorem; the theorem is a base layer rather than the whole result.
- uses
- The paper appears to rely on the theorem as machinery.
- contradicts
- The paper's claim conflicts with a theorem or certificate in the canon.
- unclear
- Pith found a possible connection, but the passage is too broad, indirect, or ambiguous to say the theorem truly supports the claim.
Reference graph
Works this paper leans on
-
[1]
J. Fang, S. Zhang, C. Wu, Z. Yang, Z. Liu, S. Li, K. Wang, W. Du, and X. Wang, “MolTC: Towards Molecular Relational Modeling In JOURNAL OF LATEX CLASS FILES, VOL. 14, NO. 8, AUGUST 2021 13 Language Models,” in Proceedings of the 62nd Annual Meeting of the Association for Computational Linguistics , 2024, pp. 1943–1958
work page 2021
-
[2]
Shift-Robust Molecular Relational Learning with Causal Substructure,
N. Lee, K. Yoon, G. S. Na, S. Kim, and C. Park, “Shift-Robust Molecular Relational Learning with Causal Substructure,” in Proceedings of the 29th ACM SIGKDD Conference on Knowledge Discovery and Data Mining, 2023, pp. 1200–1212
work page 2023
-
[3]
Individual and Structural Graph Information Bottlenecks for Out-of-Distribution Generalization,
L. Yang, J. Zheng, H. Wang, Z. Liu, Z. Huang, S. Hong, W. Zhang, and B. Cui, “Individual and Structural Graph Information Bottlenecks for Out-of-Distribution Generalization,” IEEE Transactions on Knowledge and Data Engineering , vol. 36, no. 2, pp. 682–693, 2024
work page 2024
-
[4]
Hago-Net: Hierarchical Geometric Massage Passing for Molecular Representation Learning,
H. Pei, T. Chen, A. Chen, H. Deng, J. Tao, P. Wang, and X. Guan, “Hago-Net: Hierarchical Geometric Massage Passing for Molecular Representation Learning,” in Proceedings of the 38th AAAI Conference on Artificial Intelligence , vol. 38, no. 13, 2024, pp. 14 572–14 580
work page 2024
-
[5]
OOD-GNN: Out-of- Distribution Generalized Graph Neural Network,
H. Li, X. Wang, Z. Zhang, and W. Zhu, “OOD-GNN: Out-of- Distribution Generalized Graph Neural Network,” in Proceedings of the 40th International Conference on Data Engineering , 2024, pp. 5681– 5682
work page 2024
-
[6]
DrugCLIP: Contrastive Protein-Molecule Representation Learning for Virtual Screening,
B. Gao, B. Qiang, H. Tan, Y . Jia, M. Ren, M. Lu, J. Liu, W.-Y . Ma, and Y . Lan, “DrugCLIP: Contrastive Protein-Molecule Representation Learning for Virtual Screening,” inProceedings of the 37th International Conference on Neural Information Processing Systems , vol. 36, 2023, pp. 44 595–44 614
work page 2023
-
[7]
Advancing Molecule Invariant Representation via Privileged Substructure Identification,
R. Wang, H. Dai, C. Yang, L. Song, and C. Shi, “Advancing Molecule Invariant Representation via Privileged Substructure Identification,” in Proceedings of the 30th ACM SIGKDD Conference on Knowledge Discovery and Data Mining , 2024, pp. 3188–3199
work page 2024
-
[8]
MMGNN: A Molecular Merged Graph Neural Network for Explainable Solvation Free Energy Prediction,
W. Du, S. Zhang, J. X. Di Wu, Z. Zhao, J. Fang, and Y . Wang, “MMGNN: A Molecular Merged Graph Neural Network for Explainable Solvation Free Energy Prediction,” in Proceedings of the Thirty-Third International Joint Conference on Artificial Intelligence, 2024, pp. 5808– 5816
work page 2024
-
[9]
The Key–Lock Theory and the Induced Fit Theory,
D. E. Koshland Jr, “The Key–Lock Theory and the Induced Fit Theory,” Angewandte Chemie International Edition in English, vol. 33, no. 23-24, pp. 2375–2378, 1995
work page 1995
-
[10]
From Intuition to AI: Evolution of Small Molecule Representations in Drug Discovery,
M. McGibbon, S. Shave, J. Dong, Y . Gao, D. R. Houston, J. Xie, Y . Yang, P. Schwaller, and V . Blay, “From Intuition to AI: Evolution of Small Molecule Representations in Drug Discovery,” Briefings in Bioinformatics, vol. 25, no. 1, p. bbad422, 2024
work page 2024
-
[11]
J. Xia, L. Zhang, X. Zhu, Y . Liu, Z. Gao, B. Hu, C. Tan, J. Zheng, S. Li, and S. Z. Li, “Understanding the Limitations of Deep Models for Molecular Property Prediction: Insights and Solutions,” Advances in Neural Information Processing Systems , vol. 36, pp. 64 774–64 792, 2023
work page 2023
-
[12]
Retuning of Hippocampal Representations During Sleep,
K. Maboudi, B. Giri, H. Miyawaki, C. Kemere, and K. Diba, “Retuning of Hippocampal Representations During Sleep,” Nature, pp. 1–9, 2024
work page 2024
-
[13]
Self-Explainable Temporal Graph Networks based on Graph Information Bottleneck,
S. Seo, S. Kim, J. Jung, Y . Lee, and C. Park, “Self-Explainable Temporal Graph Networks based on Graph Information Bottleneck,” in Proceedings of the 30th ACM SIGKDD Conference on Knowledge Discovery and Data Mining , 2024
work page 2024
-
[14]
J. Born and M. Manica, “Regression Transformer Enables Concurrent Sequence Regression and Generation for Molecular Language Mod- elling,” Nature Machine Intelligence , vol. 5, no. 4, pp. 432–444, 2023
work page 2023
-
[15]
Conditional Graph Information Bottleneck for Molecular Relational Learning,
N. Lee, D. Hyun, G. S. Na, S. Kim, J. Lee, and C. Park, “Conditional Graph Information Bottleneck for Molecular Relational Learning,” in Proceedings of the 40th International Conference on Machine Learning , vol. 202, 2023, pp. 18 852–18 871
work page 2023
-
[16]
Z. Li, S. Zhu, B. Shao, X. Zeng, T. Wang, and T.-Y . Liu, “DSN-DDI: An Accurate and Generalized Framework for Drug–Drug Interaction Prediction by Dual-View Representation Learning,” Briefings in Bioin- formatics, vol. 24, no. 1, p. bbac597, 2023
work page 2023
-
[17]
Geometry-Enhanced Molecular Representation Learning for Property Prediction,
X. Fang, L. Liu, J. Lei, D. He, S. Zhang, J. Zhou, F. Wang, H. Wu, and H. Wang, “Geometry-Enhanced Molecular Representation Learning for Property Prediction,” Nature Machine Intelligence, vol. 4, no. 2, pp. 127–134, 2022
work page 2022
-
[18]
J. Rao, J. Xie, Q. Yuan, D. Liu, Z. Wang, Y . Lu, S. Zheng, and Y . Yang, “A Variational Expectation-Maximization Framework for Bal- anced Multi-Scale Learning of Protein and Drug Interactions,” Nature Communications, vol. 15, no. 1, p. 4476, 2024
work page 2024
-
[19]
S.-W. Li, L.-C. Xu, C. Zhang, S.-Q. Zhang, and X. Hong, “Reac- tion Performance Prediction with an Extrapolative and Interpretable Graph Model Based on Chemical Knowledge,” Nature Communications, vol. 14, no. 1, p. 3569, 2023
work page 2023
-
[20]
N. Yang, K. Zeng, Q. Wu, and J. Yan, “Molerec: Combinatorial Drug Recommendation with Substructure-Aware Molecular Representation Learning,” in Proceedings of the 32nd ACM on Web Conference , 2023, pp. 4075–4085
work page 2023
-
[21]
Z. Yang, W. Zhong, Q. Lv, and C. Y .-C. Chen, “Learning Size-Adaptive Molecular Substructures for Explainable Drug–Drug Interaction Predic- tion by Substructure-Aware Graph Neural Network,” Chemical Science, vol. 13, no. 29, pp. 8693–8703, 2022
work page 2022
-
[22]
Z. Yang, W. Zhong, Q. Lv, T. Dong, G. Chen, and C. Y .-C. Chen, “Interaction-Based Inductive Bias in Graph Neural Networks: Enhancing Protein-Ligand Binding Affinity Predictions From 3D Structures,” IEEE Transactions on Pattern Analysis and Machine Intelligence , 2024
work page 2024
-
[23]
Artificial Intelligence in Drug Discovery and Development,
K.-K. Mak, Y .-H. Wong, and M. R. Pichika, “Artificial Intelligence in Drug Discovery and Development,” Drug Discovery and Evaluation: Safety and Pharmacokinetic Assays , pp. 1461–1498, 2024
work page 2024
-
[24]
Recent Advances in Scaffold Hopping: Miniperspective,
Y . Hu, D. Stumpfe, and J. Bajorath, “Recent Advances in Scaffold Hopping: Miniperspective,” Journal of Medicinal Chemistry , vol. 60, no. 4, pp. 1238–1246, 2017
work page 2017
-
[25]
H.-J. B ¨ohm, A. Flohr, and M. Stahl, “Scaffold Hopping,” Drug Discov- ery Today: Technologies, vol. 1, no. 3, pp. 217–224, 2004
work page 2004
-
[26]
Multi-View Graph Contrastive Representation Learning for Drug-Drug Interaction Prediction,
Y . Wang, Y . Min, X. Chen, and J. Wu, “Multi-View Graph Contrastive Representation Learning for Drug-Drug Interaction Prediction,” in Pro- ceedings of the 30th ACM on Web Conference , 2021, pp. 2921–2933
work page 2021
-
[27]
X. Su, P. Hu, Z.-H. You, S. Y . Philip, and L. Hu, “Dual-Channel Learning Framework for Drug-Drug Interaction Prediction via Relation-Aware Heterogeneous Graph Transformer,” in Proceedings of the 38th AAAI Conference on Artificial Intelligence, vol. 38, no. 1, 2024, pp. 249–256
work page 2024
-
[28]
Towards Scalable Automated Alignment of LLMs: A Survey,
B. Cao, K. Lu, X. Lu, J. Chen, M. Ren, H. Xiang, P. Liu, Y . Lu, B. He, X. Han et al. , “Towards Scalable Automated Alignment of LLMs: A Survey,” arXiv preprint arXiv:2406.01252 , 2024
-
[29]
Structure Determination of High-Energy States in a Dynamic Protein Ensemble,
J. B. Stiller, R. Otten, D. H ¨aussinger, P. S. Rieder, D. L. Theobald, and D. Kern, “Structure Determination of High-Energy States in a Dynamic Protein Ensemble,” Nature, vol. 603, no. 7901, pp. 528–535, 2022
work page 2022
-
[30]
Heterogeneous Causal Metapath Graph Neural Network for Gene- Microbe-Disease Association Prediction,
K. Zhang, F. Huang, L. Liu, Z. Xiong, H. Zhang, Y . Quan, and W. Zhang, “Heterogeneous Causal Metapath Graph Neural Network for Gene- Microbe-Disease Association Prediction,” in Proceedings of the 33rd International Joint Conference on Artificial Intelligence , 2024
work page 2024
-
[31]
D. Weininger, “SMILES, a Chemical Language and Information Sys- tem. 1. Introduction to Methodology and Encoding Rules,” Journal of Chemical Information and Computer Sciences , vol. 28, no. 1, p. 31–36, 1988
work page 1988
-
[32]
Brown, In Silico Medicinal Chemistry: Computational Methods to Support Drug Design
N. Brown, In Silico Medicinal Chemistry: Computational Methods to Support Drug Design . Royal Society of Chemistry, 2015
work page 2015
-
[33]
How Powerful are Graph Neural Networks?
K. Xu, W. Hu, J. Leskovec, and S. Jegelka, “How Powerful are Graph Neural Networks?” in Proceedings of the 7th International Conference on Learning Representations , 2019
work page 2019
-
[34]
Neural Message Passing for Quantum Chemistry,
J. Gilmer, S. S. Schoenholz, P. F. Riley, O. Vinyals, and G. E. Dahl, “Neural Message Passing for Quantum Chemistry,” in Proceedings of the 34th International Conference on Machine Learning , 2017, p. 1263–1272
work page 2017
-
[35]
P. Veli ˇckovi´c, G. Cucurull, A. Casanova, A. Romero, P. Lio, and Y . Bengio, “Graph Attention Networks,” in Proceedings of the 6th International Conference on Learning Representations , 2018
work page 2018
-
[36]
Semi-Supervised Classification with Graph Convolutional Networks,
T. N. Kipf and M. Welling, “Semi-Supervised Classification with Graph Convolutional Networks,” in Proceedings of the 5th International Con- ference on Learning Representations , 2017
work page 2017
-
[37]
A Survey on Information Bottleneck,
S. Hu, Z. Lou, X. Yan, and Y . Ye, “A Survey on Information Bottleneck,” IEEE Transactions on Pattern Analysis and Machine Intelligence , 2024
work page 2024
-
[38]
Self-Explainable Temporal Graph Networks based on Graph Information Bottleneck,
S. Seo, S. Kim, J. Jung, Y . Lee, and C. Park, “Self-Explainable Temporal Graph Networks based on Graph Information Bottleneck,” in Proceedings of the 30th ACM SIGKDD Conference on Knowledge Discovery and Data Mining , ser. KDD ’24, 2024, p. 2572–2583
work page 2024
-
[39]
Molecular Set Representation Learning,
M. Boulougouri, P. Vandergheynst, and D. Probst, “Molecular Set Representation Learning,” Nature Machine Intelligence, vol. 6, pp. 754– 763, 2024
work page 2024
-
[40]
Experimental Database of Optical Properties of Organic Compounds,
J. F. Joung, M. Han, M. Jeong, and S. Park, “Experimental Database of Optical Properties of Organic Compounds,” Scientific Data, vol. 7, no. 1, p. 295, 2020
work page 2020
-
[41]
Minnesota Solvation Database (MNSOL) Version 2012,
A. V . Marenich, C. P. Kelly, J. D. Thompson, G. D. Hawkins, C. C. Chambers, D. J. Giesen, P. Winget, C. J. Cramer, and D. G. Truhlar, “Minnesota Solvation Database (MNSOL) Version 2012,” 2020
work page 2012
-
[42]
FreeSolv: A Database of Experimental and Calculated Hydration Free Energies, with Input Files,
D. L. Mobley and J. P. Guthrie, “FreeSolv: A Database of Experimental and Calculated Hydration Free Energies, with Input Files,” Journal of Computer-Aided Molecular Design , vol. 28, pp. 711–720, 2014
work page 2014
-
[43]
E. Moine, R. Privat, B. Sirjean, and J.-N. Jaubert, “Estimation of Solva- tion Quantities from Experimental Thermodynamic Data: Development of the Comprehensive CompSol Databank for Pure and Mixed Solutes,” Journal of Physical and Chemical Reference Data , vol. 46, no. 3, 2017
work page 2017
-
[44]
L. M. Grubbs, M. Saifullah, E. Nohelli, S. Ye, S. S. Achi, W. E. Acree Jr, and M. H. Abraham, “Mathematical Correlations for Describing Solute Transfer into Functionalized Alkane Solvents Containing Hydroxyl, JOURNAL OF LATEX CLASS FILES, VOL. 14, NO. 8, AUGUST 2021 14 Ether, Ester or Ketone Solvents,”Fluid Phase Equilibria, vol. 298, no. 1, pp. 48–53, 2010
work page 2021
-
[45]
Transfer Learning for Solvation Free Energies: From Quantum Chemistry to Experiments,
F. H. Vermeire and W. H. Green, “Transfer Learning for Solvation Free Energies: From Quantum Chemistry to Experiments,” Chemical Engineering Journal, vol. 418, p. 129307, 2021
work page 2021
-
[46]
W. Zhang, Y . Chen, F. Liu, F. Luo, G. Tian, and X. Li, “Predicting Potential Drug-Drug Interactions by Integrating Chemical, Biological, Phenotypic and Network Data,” BMC Bioinformatics, vol. 18, pp. 1–12, 2017
work page 2017
-
[47]
D. S. Himmelstein and S. E. Baranzini, “Heterogeneous Network Edge Prediction: A Data Integration Approach to Prioritize Disease- Associated Genes,” PLoS Computational Biology , vol. 11, no. 7, p. e1004259, 2015
work page 2015
-
[48]
DrugBank 5.0: A Major Update to the DrugBank Database for 2018,
D. S. Wishart, Y . D. Feunang, A. C. Guo, E. J. Lo, A. Marcu, J. R. Grant, T. Sajed, D. Johnson, C. Li, Z. Sayeeda et al. , “DrugBank 5.0: A Major Update to the DrugBank Database for 2018,” Nucleic Acids Research, vol. 46, no. D1, pp. D1074–D1082, 2018
work page 2018
-
[49]
Multilevel Algorithms for Multi-Constraint Graph Partitioning,
G. Karypis and V . Kumar, “Multilevel Algorithms for Multi-Constraint Graph Partitioning,” in Proceedings of the ACM/IEEE Conference on Supercomputing, 1998, pp. 28–28
work page 1998
-
[50]
Y . Pathak, S. Laghuvarapu, S. Mehta, and U. D. Priyakumar, “Chem- ically Interpretable Graph Interaction Network for Prediction of Phar- macokinetic Properties of Drug-Like Molecules,” in Proceedings of the 34th AAAI Conference on Artificial Intelligence , vol. 34, no. 01, 2020, pp. 873–880
work page 2020
-
[51]
SSI–DDI: Substructure– Substructure Interactions for Drug–Drug Interaction Prediction,
A. K. Nyamabo, H. Yu, and J.-Y . Shi, “SSI–DDI: Substructure– Substructure Interactions for Drug–Drug Interaction Prediction,” Brief- ings in Bioinformatics , vol. 22, no. 6, p. bbab133, 2021
work page 2021
-
[52]
Mantel Test in Population Genetics,
J. A. F. Diniz-Filho, T. N. Soares, J. S. Lima, R. Dobrovolski, V . L. Landeiro, M. P. d. C. Telles, T. F. Rangel, and L. M. Bini, “Mantel Test in Population Genetics,” Genetics and Molecular Biology , vol. 36, pp. 475–485, 2013
work page 2013
-
[53]
Uncovering Neural Scaling Laws in Molecular Representation Learning,
D. Chen, Y . Zhu, J. Zhang, Y . Du, Z. Li, Q. Liu, S. Wu, and L. Wang, “Uncovering Neural Scaling Laws in Molecular Representation Learning,” Advances in Neural Information Processing Systems, vol. 36, 2024
work page 2024
-
[54]
D. Wu, W. Sun, Y . He, Z. Chen, and X. Luo, “MKG-FENN: A Multimodal Knowledge Graph Fused End-to-End Neural Network for Accurate Drug–Drug Interaction Prediction,” in Proceedings of the 38th AAAI Conference on Artificial Intelligence , vol. 38, no. 9, 2024, pp. 10 216–10 224
work page 2024
-
[55]
DDIPrompt: Drug-Drug Interaction Event Prediction based on Graph Prompt Learning,
Y . Wang, Y . Xiong, X. Wu, X. Sun, J. Zhang, and G. Zheng, “DDIPrompt: Drug-Drug Interaction Event Prediction based on Graph Prompt Learning,” in Proceedings of the 33rd ACM International Conference on Information and Knowledge Management , ser. CIKM ’24. New York, NY , USA: Association for Computing Machinery, 2024, p. 2431–2441
work page 2024
-
[56]
A Molecular Video-Derived Foundation Model for Scientific Drug Discovery,
H. Xiang, L. Zeng, L. Hou, K. Li, Z. Fu, Y . Qiu, R. Nussinov, J. Hu, M. Rosen-Zvi, X. Zeng et al., “A Molecular Video-Derived Foundation Model for Scientific Drug Discovery,” Nature Communications, vol. 15, no. 1, p. 9696, 2024
work page 2024
-
[57]
Learning Motif-Based Graphs for Drug–Drug Interaction Prediction via Local–Global Self-Attention,
Y . Zhong, G. Li, J. Yang, H. Zheng, Y . Yu, J. Zhang, H. Luo, B. Wang, and Z. Weng, “Learning Motif-Based Graphs for Drug–Drug Interaction Prediction via Local–Global Self-Attention,”Nature Machine Intelligence, vol. 6, no. 9, pp. 1094–1105, 2024
work page 2024
-
[58]
R. Zhang, X. Wang, P. Wang, Z. Meng, W. Cui, and Y . Zhou, “HTCL- DDI: A Hierarchical Triple-view Contrastive Learning Framework for Drug–Drug Interaction Prediction,” Briefings in Bioinformatics, vol. 24, no. 6, p. bbad324, 09 2023
work page 2023
-
[59]
Lima: Less is More for Alignment,
C. Zhou, P. Liu, P. Xu, S. Iyer, J. Sun, Y . Mao, X. Ma, A. Efrat, P. Yu, L. Yu et al., “Lima: Less is More for Alignment,” Advances in Neural Information Processing Systems , vol. 36, 2024
work page 2024
-
[60]
Ope- nassistant Conversations-Democratizing Large Language Model Align- ment,
A. K ¨opf, Y . Kilcher, D. von R ¨utte, S. Anagnostidis, Z. R. Tam, K. Stevens, A. Barhoum, D. Nguyen, O. Stanley, R. Nagyfi et al., “Ope- nassistant Conversations-Democratizing Large Language Model Align- ment,” Advances in Neural Information Processing Systems , vol. 36, 2024
work page 2024
-
[61]
Beavertails: Towards Improved Safety Align- ment of LLM via a Human-Preference Dataset,
J. Ji, M. Liu, J. Dai, X. Pan, C. Zhang, C. Bian, B. Chen, R. Sun, Y . Wang, and Y . Yang, “Beavertails: Towards Improved Safety Align- ment of LLM via a Human-Preference Dataset,” Advances in Neural Information Processing Systems , vol. 36, 2024
work page 2024
-
[62]
H. R. Kirk, B. Vidgen, P. R ¨ottger, and S. A. Hale, “The Benefits, Risks and Bounds of Personalizing the Alignment of Large Language Models to Individuals,” Nature Machine Intelligence , pp. 1–10, 2024
work page 2024
-
[63]
Protein Remote Homology Detection and Structural Alignment Using Deep Learning,
T. Hamamsy, J. T. Morton, R. Blackwell, D. Berenberg, N. Carriero, V . Gligorijevic, C. E. Strauss, J. K. Leman, K. Cho, and R. Bonneau, “Protein Remote Homology Detection and Structural Alignment Using Deep Learning,” Nature Biotechnology , vol. 42, no. 6, pp. 975–985, 2024
work page 2024
-
[64]
Y . Wang, D. Wang, H. Liu, B. Hu, Y . Yan, Q. Zhang, and Z. Zhang, “Optimizing Long-tailed Link Prediction in Graph Neural Networks through Structure Representation Enhancement,” in Proceedings of the 30th ACM SIGKDD Conference on Knowledge Discovery and Data Mining, 2024, pp. 3222–3232
work page 2024
-
[65]
Graphormer Supervised De Novo Protein Design Method and Function Validation,
J. Mu, Z. Li, B. Zhang, Q. Zhang, J. Iqbal, A. Wadood, T. Wei, Y . Feng, and H.-F. Chen, “Graphormer Supervised De Novo Protein Design Method and Function Validation,” Briefings in Bioinformatics , vol. 25, no. 3, p. bbae135, 2024
work page 2024
-
[66]
DGCL: Dual-Graph Neural Networks Contrastive Learning for Molecular Property Prediction,
X. Jiang, L. Tan, and Q. Zou, “DGCL: Dual-Graph Neural Networks Contrastive Learning for Molecular Property Prediction,” Briefings in Bioinformatics, vol. 25, no. 6, p. bbae474, 2024. Peiliang Zhang (Student Member, IEEE) is pur- suing his Ph.D. degree at Wuhan University of Technology, Wuhan, China. He is also currently a visiting Ph.D. student at Yonsei...
work page 2024
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.