Modeling phase separation in polymer-derived silicon carbonitride ceramics through extended machine learning molecular dynamics
Pith reviewed 2026-05-21 07:24 UTC · model grok-4.3
The pith
Machine learning molecular dynamics reveals carbon nucleating into graphene-like sheets within the SiCN ceramic matrix during heating.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
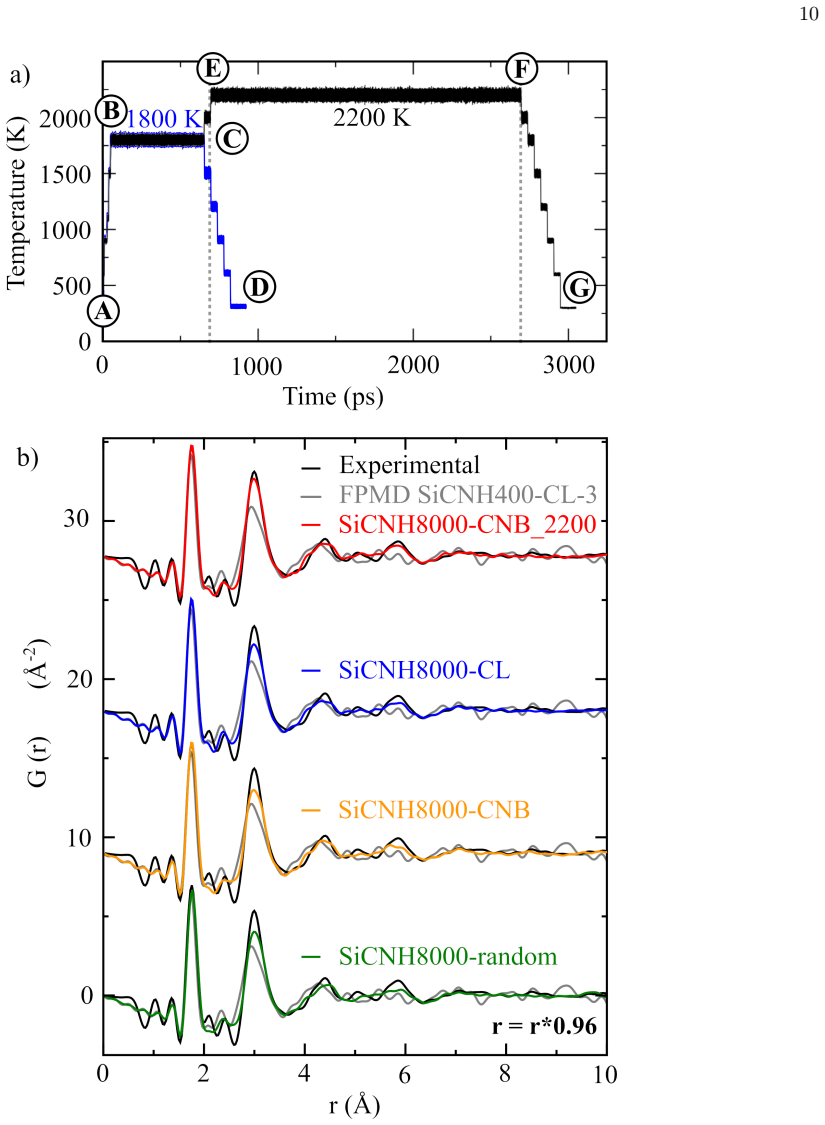
The central discovery is a phase separation process in which carbon domains nucleate from the amorphous SiCN matrix during thermal treatment, forming graphene-like sheets while the integrity of the ceramic network is preserved. Defective five- and seven-membered carbon rings mediate the conversion to stable six-membered aromatic structures. The simulated atomic configurations reproduce experimental pair distribution functions with high fidelity, supplying atomic-scale explanations for the combination of ceramic thermal stability and graphitic features in these materials.
What carries the argument
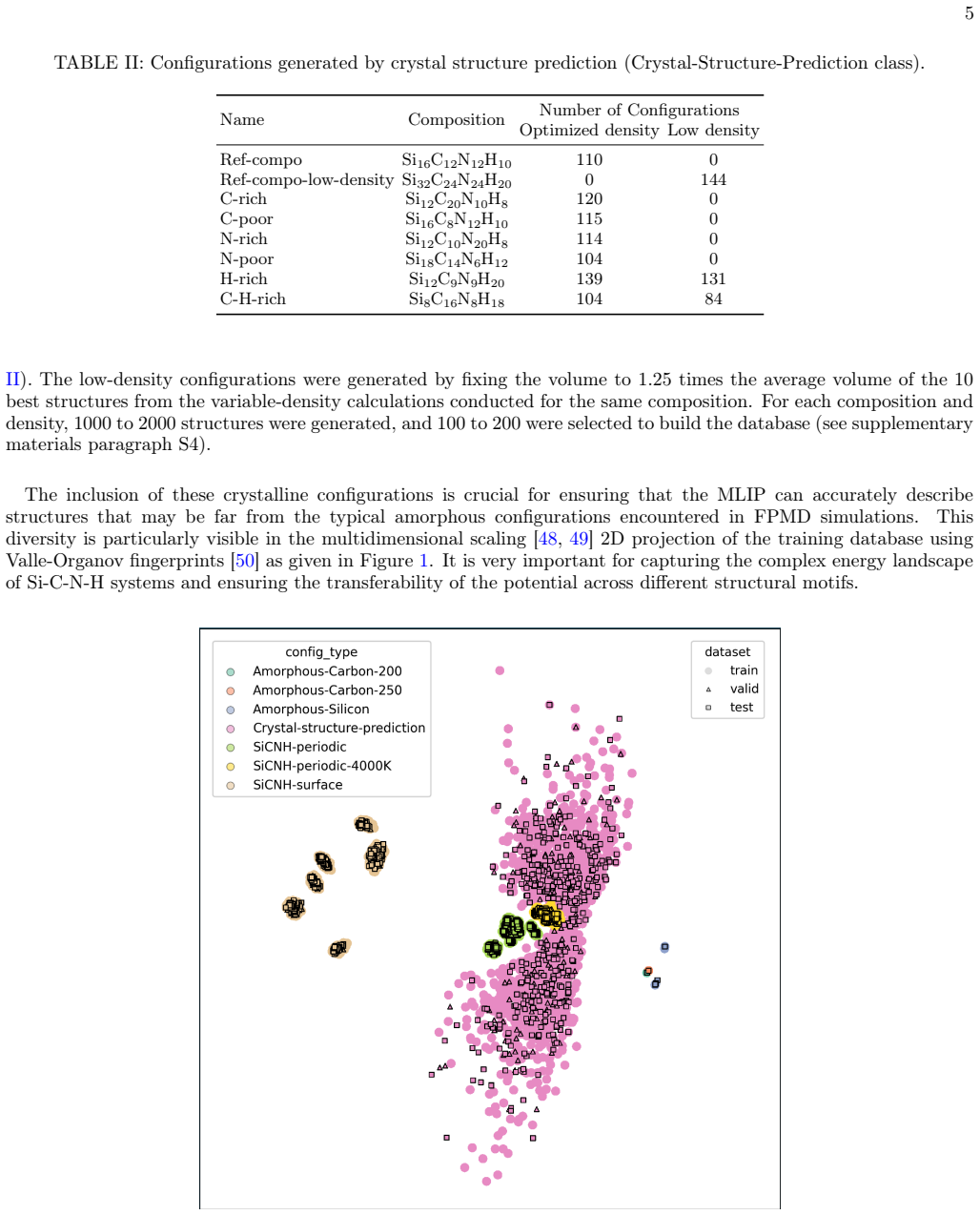
A machine learning interatomic potential trained on a diversified database of over 9000 configurations that enables large-scale molecular dynamics simulations of 8000-atom SiCNH systems at high temperature.
If this is right
- The method supplies atomic-level mechanisms for the thermal stability and mixed properties of polymer-derived ceramics.
- Defective carbon rings are identified as the structural intermediates that enable the transition to ordered graphitic domains.
- The same potential framework can be applied to other complex amorphous multicomponent systems at experimentally relevant sizes.
- Microscopic pathways for structural transformation during processing are now accessible for direct comparison with experiment.
Where Pith is reading between the lines
- Processing temperatures or times could be adjusted to control the size and connectivity of the emerging carbon sheets for targeted electrical or mechanical performance.
- The same simulation approach might be extended to predict how dopants or different polymer precursors alter the phase-separation route.
- Longer simulation timescales could reveal whether the graphene-like domains continue to grow or stabilize once formed.
Load-bearing premise
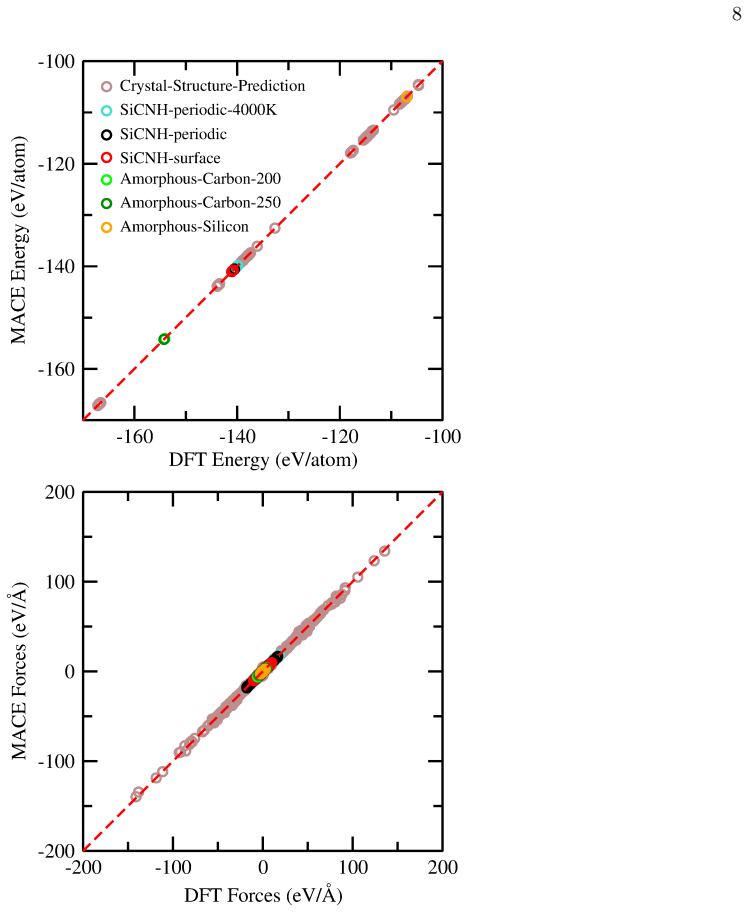
The trained machine learning potential reproduces the energies and dynamics of carbon nucleation and ring changes in large systems without large extrapolation errors or missing interactions.
What would settle it
A simulation run at the same temperature and scale that produces no carbon domain nucleation or that yields atomic pair distribution functions clearly different from experimental measurements would falsify the reported phase-separation mechanism.
Figures





read the original abstract
Polymer-derived ceramics combine the thermal stability of ceramics with the versatile properties of carbon domains, but modeling their atomic-scale evolution during processing remains elusive due to the limitations of traditional computational methods. To address this issue, here we develop and apply a machine learning interatomic potential for silicon carbonitride-based (Si-C-N-H) systems, trained on a diversified database of over 9000 configurations -- including amorphous models, high-temperature states, surfaces, and crystal structure predictions -- to capture the full complexity of these materials. This potential enables large-scale molecular dynamics simulations of 8000-atom systems revealing the atomic-scale evolution of the polymer-derived ceramic during thermal treatment. A key result of this work is the occurrence of a phase separation where carbon domains progressively nucleate from the amorphous SiCN matrix during thermal processing, forming distinct graphene-like sheets while preserving the integrity of the ceramic network. The resulting models reproduce the experimental atomic pair distribution functions with exceptional fidelity, validating our approach and providing microscopic explanations for the material unique combination of ceramic and graphitic properties. In this process, defective 5- and/or 7-member carbon rings, mediate the transformation to stable 6-member aromatic structures. These findings offer new atomic-scale insights into the thermal stability and structural transformation pathways of polymer-derived ceramics, while our methodology opens avenues for studying complex amorphous systems with first-principles accuracy at experimentally relevant scales.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The paper develops a machine learning interatomic potential for Si-C-N-H systems trained on a diversified database of over 9000 configurations (including amorphous, high-temperature, surface, and crystal structures). It applies this potential to large-scale MD simulations of 8000-atom polymer-derived SiCN models during thermal processing. The central claim is that carbon domains nucleate from the amorphous matrix to form graphene-like sheets via mediation by defective 5- and 7-membered rings that rearrange into stable 6-membered aromatic structures, while the final atomic configurations reproduce experimental pair distribution functions with high fidelity.
Significance. If the dynamical pathway is faithfully reproduced, the work provides valuable atomic-scale mechanistic insight into phase separation and carbon domain formation in polymer-derived ceramics, explaining their hybrid ceramic-graphitic properties at experimentally relevant scales. The scale-up to 8000-atom systems and reproduction of experimental PDFs are notable strengths that could inform processing strategies.
major comments (1)
- [MD simulation results and validation section] The load-bearing claim is that the ML potential correctly drives carbon nucleation and the 5-/7- to 6-membered ring conversion during high-T annealing. However, validation is reported only via reproduction of experimental PDFs for the final structures (Abstract and results on MD trajectories). PDF agreement constrains equilibrium pair correlations but does not test relative energies or barriers of the defective-ring intermediates identified as mediating the transformation, leaving open the possibility that the observed pathway is an extrapolation artifact rather than a physically faithful mechanism.
minor comments (2)
- [Methods] The training database is described at a high level ('diversified database of over 9000 configurations'); more detail on the distribution of high-temperature states and how extrapolation risk was quantified for the 8000-atom runs would strengthen the methods.
- [Results] Error bars or uncertainty estimates on the PDF comparisons and on the reported ring statistics during annealing are not mentioned; adding these would improve clarity of the validation.
Simulated Author's Rebuttal
We thank the referee for the detailed and constructive review of our manuscript. We address the major comment regarding validation of the dynamical pathway below, and we outline revisions that will strengthen the presentation of our results.
read point-by-point responses
-
Referee: [MD simulation results and validation section] The load-bearing claim is that the ML potential correctly drives carbon nucleation and the 5-/7- to 6-membered ring conversion during high-T annealing. However, validation is reported only via reproduction of experimental PDFs for the final structures (Abstract and results on MD trajectories). PDF agreement constrains equilibrium pair correlations but does not test relative energies or barriers of the defective-ring intermediates identified as mediating the transformation, leaving open the possibility that the observed pathway is an extrapolation artifact rather than a physically faithful mechanism.
Authors: We agree that reproduction of experimental PDFs validates the final atomic configurations but does not directly probe the relative energies or barriers associated with the 5-/7- to 6-membered ring rearrangements. Our ML potential was trained on a diversified database of over 9000 configurations that explicitly includes high-temperature amorphous states, surfaces, and crystal structures, which encompass a broad range of carbon ring environments and defective motifs. This training strategy is intended to ensure faithful description of the relevant energetics. Nevertheless, to directly address the concern about possible extrapolation artifacts, the revised manuscript will include new analysis: we will report relative energies (computed with both the ML potential and reference DFT) for representative small-model systems containing 5-, 6-, and 7-membered carbon rings and their interconversions. These additional checks will confirm that the observed mediation pathway is consistent with the underlying potential energy surface learned from the training data. revision: yes
Circularity Check
No significant circularity; derivation relies on external training data and experimental validation
full rationale
The paper trains a machine learning interatomic potential on an external diversified database of over 9000 configurations (including amorphous models and high-temperature states) and then performs large-scale MD simulations on 8000-atom systems. The reported phase separation and ring rearrangements emerge as simulation outcomes, with final structures validated against independent experimental PDFs. No equations, fitted parameters, or self-citations reduce the central claim to the inputs by construction; the workflow remains self-contained against external benchmarks.
Axiom & Free-Parameter Ledger
free parameters (1)
- ML interatomic potential hyperparameters
axioms (1)
- domain assumption The diversified training database of over 9000 configurations sufficiently samples the relevant configuration space for amorphous, high-temperature, and surface states in Si-C-N-H systems.
Lean theorems connected to this paper
-
IndisputableMonolith/Cost/FunctionalEquation.leanwashburn_uniqueness_aczel unclear?
unclearRelation between the paper passage and the cited Recognition theorem.
defective 5- and/or 7-member carbon rings mediate the transformation to stable 6-member aromatic structures
-
IndisputableMonolith/Foundation/BranchSelection.leanbranch_selection unclear?
unclearRelation between the paper passage and the cited Recognition theorem.
phase separation where carbon domains progressively nucleate from the amorphous SiCN matrix
What do these tags mean?
- matches
- The paper's claim is directly supported by a theorem in the formal canon.
- supports
- The theorem supports part of the paper's argument, but the paper may add assumptions or extra steps.
- extends
- The paper goes beyond the formal theorem; the theorem is a base layer rather than the whole result.
- uses
- The paper appears to rely on the theorem as machinery.
- contradicts
- The paper's claim conflicts with a theorem or certificate in the canon.
- unclear
- Pith found a possible connection, but the passage is too broad, indirect, or ambiguous to say the theorem truly supports the claim.
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.