Prototype-Guided Latent Alignment for Data-Efficient Fine-Tuning of Molecular Foundation Models
Pith reviewed 2026-06-29 06:39 UTC · model grok-4.3
The pith
Prototype-guided alignment of latent atom representations improves low-data fine-tuning of molecular foundation models by anchoring energies to source prototypes.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
By identifying local structural similarities between source and target domains through clustering of latent representations from the pretrained model, and aligning each target-domain atom's energy contribution to its corresponding source-domain prototype, the approach introduces an inductive bias that anchors the fine-tuned model to the pretrained structure, enabling effective reuse of learned interactions and improved generalization in data-scarce regimes without restrictive assumptions on target chemistry.
What carries the argument
Prototype-guided latent alignment, which clusters atoms by their latent representations to define source prototypes and aligns target atom energies to those prototypes.
Load-bearing premise
Local structural similarities captured in the pretrained latent space reliably indicate which energy contributions can be transferred from source to target without distorting predictions on chemically diverse systems.
What would settle it
A controlled test on a target dataset with no structural overlap in latent space where the prototype alignment increases energy prediction error relative to plain fine-tuning.
Figures

read the original abstract
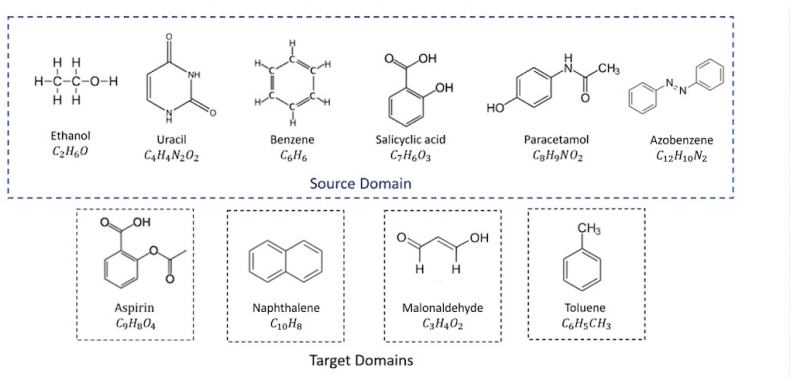
Machine learning interatomic potentials (MLIPs) have transformed materials discovery by leveraging graph neural networks (GNNs) to predict material properties with near density functional theory (DFT) accuracy. While large-scale pretrained foundation models offer transferable baseline representations, they frequently struggle to generalise to out-of-distribution (OOD) target systems -- a common challenge in modelling complex or chemically diverse materials. Fine-tuning is the standard remedy, but the high cost of generating DFT-labelled configurations confines adaptation to data-scarce regimes, where over-parameterised GNNs amplify overfitting and degrade target-domain performance. To address this, we propose a prototype-based alignment approach for data-efficient fine-tuning of MLIPs. Our method identifies local structural similarities between the source and target domains by grouping atoms with analogous chemical environments based on their latent representations. Each target-domain atom's energy contribution is aligned to its source-domain prototype, introducing an inductive bias that anchors fine-tuned representations to the pretrained structure, encouraging effective reuse of learned interactions and improving generalisation without restrictive assumptions on the target chemistry. We evaluate our method on the rMD17 benchmark using equivariant MACE and invariant SchNet across varying data budgets, and extend evaluation to the MACE-OFF foundation models on the SPICE dataset. Our approach consistently improves predictive accuracy in the low-data regime, reducing energy MAE by up to 18% over standard fine-tuning baselines.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The paper proposes a prototype-guided latent alignment method for data-efficient fine-tuning of molecular foundation models (e.g., equivariant MACE, invariant SchNet, and MACE-OFF). It groups atoms by similar chemical environments in the pretrained latent space, aligns target-domain per-atom energies to source-domain prototypes, and claims this supplies a useful inductive bias that improves generalization on OOD systems without restrictive assumptions on target chemistry. Experiments on rMD17 across data budgets and on SPICE report consistent gains, with energy MAE reductions of up to 18% versus standard fine-tuning baselines.
Significance. If the central claim holds, the approach would address a practical bottleneck in adapting large MLIPs to chemically diverse or OOD materials under tight DFT data budgets. The evaluation on established benchmarks (rMD17, SPICE) and multiple architectures is a positive feature; reproducible gains in the low-data regime would be of clear interest to the MLIP community.
major comments (2)
- [Abstract] Abstract (method description paragraph): the claim that prototype alignment supplies a net-positive inductive bias 'without restrictive assumptions on the target chemistry' is load-bearing for the central result, yet the manuscript provides no direct test that latent-nearest-neighbor atoms across domains have comparable DFT energy contributions. If source and target atoms matched by latent distance systematically differ in per-atom energy, the added alignment term becomes a source of bias rather than regularization, undermining the reported MAE reductions.
- [Evaluation sections] Evaluation sections (rMD17 and SPICE results): the headline 18% MAE improvement is reported only as aggregate numbers; without per-system breakdowns, error bars across multiple random seeds, or an ablation that isolates the alignment loss from other fine-tuning choices, it is impossible to confirm that the gains arise from the proposed inductive bias rather than from hyper-parameter differences or data-split artifacts.
minor comments (2)
- [Method] Notation for the alignment loss and prototype selection procedure should be formalized with an equation; the current prose description leaves the precise weighting between the alignment term and the standard energy/force loss unclear.
- [Abstract] The abstract states evaluation 'across varying data budgets' but does not specify the exact budgets or the number of independent runs; adding a table with these details would improve reproducibility.
Simulated Author's Rebuttal
We thank the referee for their constructive comments on our manuscript. We address each major comment below and outline the revisions we will make to strengthen the paper.
read point-by-point responses
-
Referee: [Abstract] Abstract (method description paragraph): the claim that prototype alignment supplies a net-positive inductive bias 'without restrictive assumptions on the target chemistry' is load-bearing for the central result, yet the manuscript provides no direct test that latent-nearest-neighbor atoms across domains have comparable DFT energy contributions. If source and target atoms matched by latent distance systematically differ in per-atom energy, the added alignment term becomes a source of bias rather than regularization, undermining the reported MAE reductions.
Authors: We acknowledge that the manuscript does not include a direct empirical check of whether latent-nearest-neighbor atoms across domains exhibit comparable per-atom DFT energies. The method is motivated by the pretrained latent space capturing chemically similar environments, but we agree that an explicit validation would better support the claim of a net-positive inductive bias. In the revised manuscript we will add an analysis (in the supplementary material) that compares the per-atom energies of source and target atoms matched by latent distance on the evaluated datasets. revision: yes
-
Referee: [Evaluation sections] Evaluation sections (rMD17 and SPICE results): the headline 18% MAE improvement is reported only as aggregate numbers; without per-system breakdowns, error bars across multiple random seeds, or an ablation that isolates the alignment loss from other fine-tuning choices, it is impossible to confirm that the gains arise from the proposed inductive bias rather than from hyper-parameter differences or data-split artifacts.
Authors: We agree that aggregate metrics alone limit interpretability. The original submission emphasized overall trends across data budgets, but we will expand the evaluation sections to include per-system MAE tables for rMD17, error bars computed over multiple random seeds, and an ablation that removes the alignment term while keeping all other fine-tuning choices fixed. These additions will be incorporated in the revised manuscript. revision: yes
Circularity Check
No circularity; empirical gains measured on external benchmarks
full rationale
The paper proposes a prototype-guided latent alignment method for data-efficient fine-tuning of MLIPs and reports empirical accuracy improvements (up to 18% MAE reduction) on standard external benchmarks including rMD17 and SPICE. No equations, derivations, or predictions are presented that reduce by construction to fitted parameters, self-definitions, or self-citation chains. The central claim rests on experimental comparison against standard fine-tuning baselines rather than any closed mathematical loop or imported uniqueness result, rendering the evaluation self-contained against independent data.
Axiom & Free-Parameter Ledger
Reference graph
Works this paper leans on
-
[1]
URL https://link.aps.org/doi/10.1103/P hysRevLett.104.136403
doi: 10.1103/PhysRevLett.104.136403. URL https://link.aps.org/doi/10.1103/P hysRevLett.104.136403. Albert P. Bartók, Risi Kondor, and Gábor Csányi. On representing chemical environments.Physical Review B, 87(18), May 2013. ISSN 1550-235X. doi: 10.1103/physrevb.87.184115. URL http://dx.doi.org/10.1103/PhysRevB.87.184115. Ilyes Batatia, David P Kovacs, Greg...
-
[2]
URLhttps://www.sciencedirect.com/science/article/pii/S0021999114008353. Laurens Van der Maaten and Geoffrey Hinton. Visualizing data using t-sne.Journal of machine learning research, 9(11), 2008. Scott I Vrieze. Model selection and psychological theory: a discussion of the differences between the akaike information criterion (aic) and the bayesian informa...
work page doi:10.1016/j 2008
-
[3]
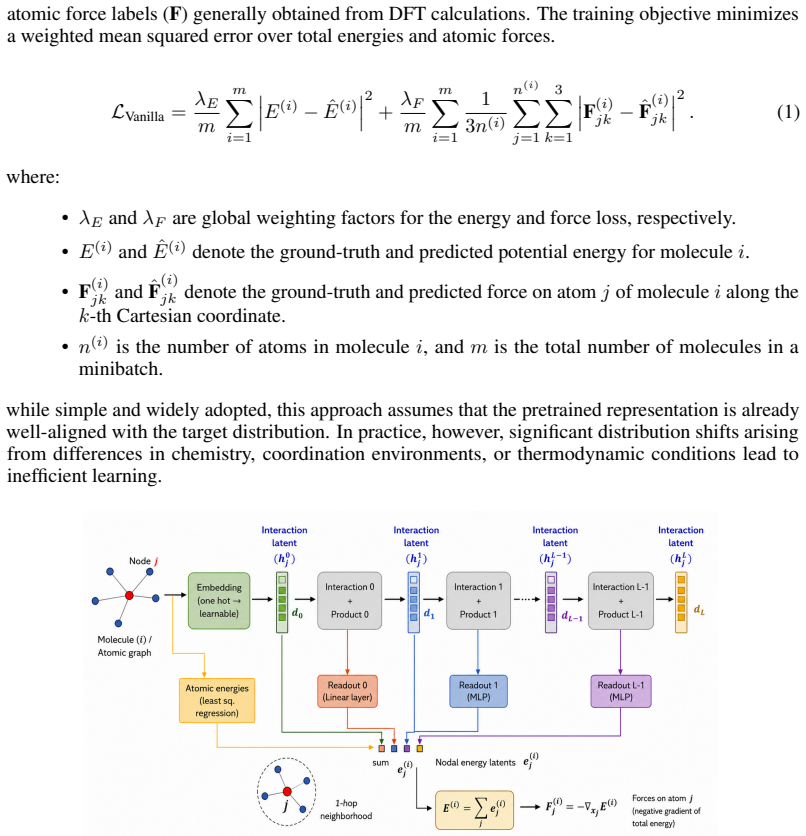
This representation captures multi-scale structural and geometric information about the atomic neighborhood
Interaction Latents:Scalar features are extracted from the outputs of Interaction0 and Interaction1 and concatenated to form the interaction latent h(i) j = Concat h(i,0) j ,h (i,1) j . This representation captures multi-scale structural and geometric information about the atomic neighborhood
-
[4]
The stacked intermediate energy contributions form the nodal energy latente(i) j , encoding task-relevant energetic information
Nodal Energy Contribution Latents:Each readout layer predicts intermediate atomic energy contributions, which are summed to obtain the final atomic energy: E(i) j = X k E(i,k) j . The stacked intermediate energy contributions form the nodal energy latente(i) j , encoding task-relevant energetic information. Concatenation of the features produced by both t...
2008
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.