Structure-Guided Adaptive Propagation for Protein-Protein Interaction Site Prediction
Pith reviewed 2026-06-28 14:28 UTC · model grok-4.3
The pith
SGAP-PPIS generates residue-wise propagation coefficients from multi-scale geometric states to let each residue adaptively balance local features and neighborhood diffusion.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
SGAP-PPIS leverages multi-scale geometric states from an equivariant graph neural network to generate residue-wise propagation coefficients, allowing each residue to adaptively balance local feature preservation and neighborhood diffusion according to its geometric microenvironment and thereby achieve competitive performance among state-of-the-art methods on Test_60.
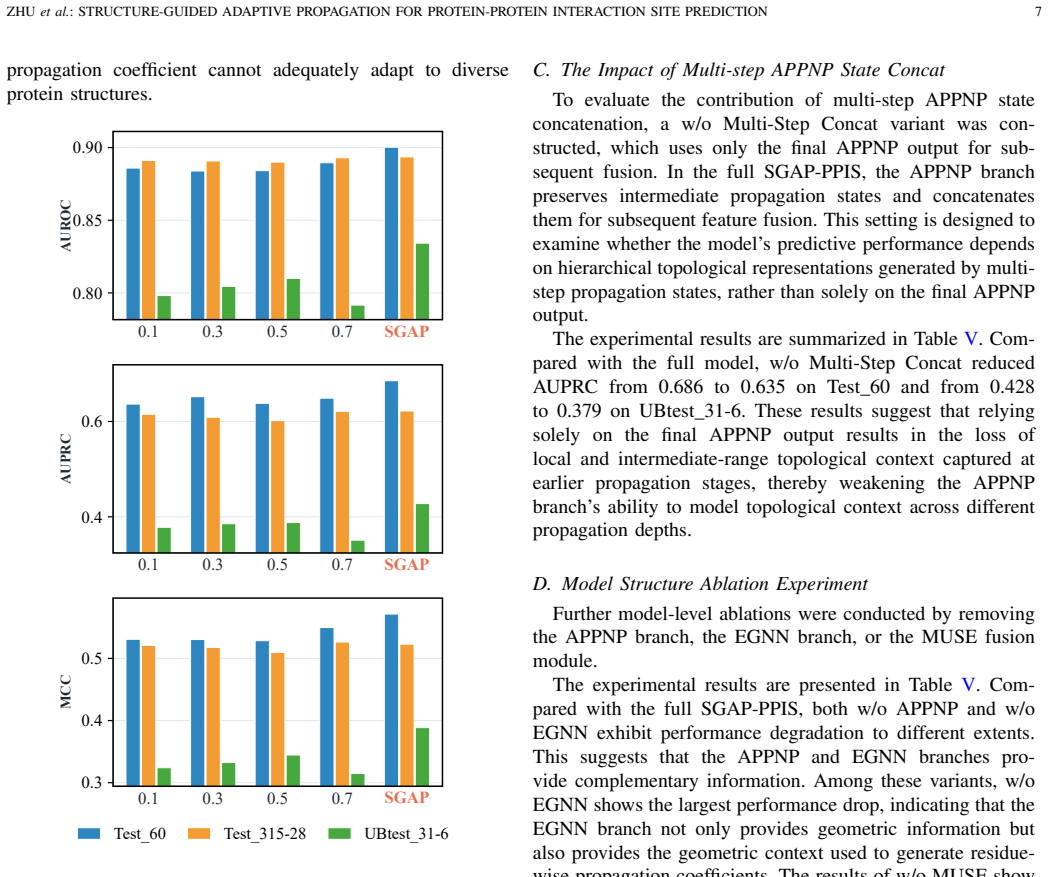
What carries the argument
Residue-wise propagation coefficients generated from multi-scale geometric states of an equivariant graph neural network.
If this is right
- True interaction sites become easier to separate from non-interacting residues that share similar local structure.
- Performance improvements arise jointly from geometry-conditioned adaptive propagation, scale-aligned geometric guidance, and multi-step propagation-state representation.
- The model maintains competitive standing with existing state-of-the-art PPIS predictors on the Test_60 set.
Where Pith is reading between the lines
- The same adaptive-coefficient idea could be tested on other graph-based tasks that predict functional sites on proteins or other biomolecules.
- If local geometry truly governs information flow at interfaces, experimental maps of interface residues might show systematic patterns tied to curvature or packing density.
- Architectures that already use equivariant networks could adopt similar residue-wise adaptation without changing the core message-passing layers.
Load-bearing premise
Fixed propagation schemes cannot distinguish true interaction sites from structurally similar non-interacting neighbors because they ignore differences in local geometric environments.
What would settle it
A controlled experiment in which a fixed-propagation baseline matches or exceeds SGAP-PPIS accuracy on Test_60 after equalizing other model components would falsify the claim that adaptive geometry-guided coefficients are necessary for the reported gains.
Figures


read the original abstract
Accurate prediction of protein-protein interaction sites (PPIS) is essential for understanding cellular processes, disease mechanisms, and therapeutic target discovery. Graph-based deep learning has advanced PPIS prediction by incorporating residue-level structural context. However, most graph-based models still rely on fixed propagation schemes that treat all residues similarly, despite the structural and functional heterogeneity of protein interfaces. Such propagation may limit the ability to adapt information diffusion to local geometric environments, making it difficult to distinguish true interaction sites from structurally similar non-interacting neighbors. We present SGAP-PPIS, a structure-guided adaptive propagation model for PPIS prediction. Rather than using a fixed propagation mechanism, SGAP-PPIS leverages multi-scale geometric states from an equivariant graph neural network to generate residue-wise propagation coefficients. This design allows each residue to adaptively balance local feature preservation and neighborhood diffusion according to its geometric microenvironment. Experimental results show that SGAP-PPIS achieves competitive performance among the state-of-the-art methods on Test\_60. Ablation studies show that geometry-conditioned adaptive propagation, scale-aligned geometric guidance, and multi-step propagation-state representation jointly drive these improvements.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript introduces SGAP-PPIS, a graph-based model for protein-protein interaction site (PPIS) prediction. It replaces fixed propagation in equivariant GNNs with residue-wise adaptive coefficients derived from multi-scale geometric states; each residue thereby balances local feature preservation against neighborhood diffusion according to its local geometry. The central empirical claim is competitive performance versus state-of-the-art methods on the Test_60 benchmark, with ablation experiments attributing the gains to geometry-conditioned adaptive propagation, scale-aligned guidance, and multi-step state representation.
Significance. If the reported performance and ablation results hold under rigorous evaluation, the work supplies a concrete mechanism for making propagation geometry-aware in PPIS models. The explicit attribution of gains to three design choices via ablation studies is a positive feature that strengthens the mechanistic interpretation.
major comments (2)
- [Abstract, §4] Abstract and §4 (Experiments): the claim of 'competitive performance among the state-of-the-art methods on Test_60' is the central empirical result, yet the abstract supplies neither numerical metrics (e.g., AUC, F1, precision-recall), error bars, nor the exact composition of Test_60. Without these quantities the strength of the claim cannot be assessed from the provided description.
- [§2, §3] §2 (Related Work) and §3 (Method): the motivating premise that fixed propagation schemes 'limit the ability to adapt information diffusion to local geometric environments' is stated without a direct quantitative demonstration (e.g., a controlled comparison showing that a non-adaptive baseline fails specifically on structurally similar non-interacting neighbors). This assumption underpins the design choice but is not load-bearing for the performance claim itself.
minor comments (2)
- [§3] Ensure that all equations defining the residue-wise propagation coefficients (likely in §3) are accompanied by explicit pseudocode or a small worked example so that the mapping from multi-scale geometric states to coefficients is fully reproducible.
- [§4] Table or figure captions in the experimental section should explicitly list the exact hyper-parameter settings and random seeds used for the reported runs.
Simulated Author's Rebuttal
We thank the referee for the constructive feedback and recommendation of minor revision. We address the major comments point by point below.
read point-by-point responses
-
Referee: [Abstract, §4] Abstract and §4 (Experiments): the claim of 'competitive performance among the state-of-the-art methods on Test_60' is the central empirical result, yet the abstract supplies neither numerical metrics (e.g., AUC, F1, precision-recall), error bars, nor the exact composition of Test_60. Without these quantities the strength of the claim cannot be assessed from the provided description.
Authors: We agree that the abstract should be self-contained. In the revision we will add the key metrics (AUC and F1 with standard deviations) achieved on Test_60 together with a brief statement of the benchmark composition. The detailed results and error bars already appear in §4. revision: yes
-
Referee: [§2, §3] §2 (Related Work) and §3 (Method): the motivating premise that fixed propagation schemes 'limit the ability to adapt information diffusion to local geometric environments' is stated without a direct quantitative demonstration (e.g., a controlled comparison showing that a non-adaptive baseline fails specifically on structurally similar non-interacting neighbors). This assumption underpins the design choice but is not load-bearing for the performance claim itself.
Authors: The premise is introduced conceptually to motivate the design. Quantitative support is supplied by the ablation studies that directly compare adaptive versus fixed propagation and attribute performance gains to the geometry-conditioned mechanism. Because the referee correctly notes that the assumption is not load-bearing for the central performance claim, we do not plan additional experiments or textual changes. revision: no
Circularity Check
No significant circularity
full rationale
The paper presents SGAP-PPIS as an empirical architecture that feeds multi-scale geometric states from an equivariant GNN into residue-wise propagation coefficients. The central claim is competitive performance on Test_60, justified by ablation studies that isolate the contribution of geometry-conditioned adaptation, scale alignment, and multi-step states. No derivation step, equation, or performance metric is shown to reduce by construction to a fitted parameter, self-citation, or renamed input; the model is built from standard equivariant GNN primitives and evaluated externally. This is the normal non-circular case for an applied architecture paper.
Axiom & Free-Parameter Ledger
Reference graph
Works this paper leans on
-
[1]
Protein–protein interactions define specificity in signal transduction,
T. Pawson and P. Nash, “Protein–protein interactions define specificity in signal transduction,”Genes & development, vol. 14, no. 9, pp. 1027– 1047, 2000
2000
-
[2]
A protein-interaction network of interferon-stimulated genes extends the innate immune system landscape,
P. Hubel, C. Urban, V . Bergant, W. M. Schneider, B. Knauer, A. Stukalov, P. Scaturro, A. Mann, L. Brunotte, H. H. Hoffmannet al., “A protein-interaction network of interferon-stimulated genes extends the innate immune system landscape,”Nature immunology, vol. 20, no. 4, pp. 493–502, 2019
2019
-
[3]
Wirtz, O
M. Wirtz, O. Berkowitz, M. Droux, and R. Hell, “The cysteine synthase complex from plants: mitochondrial serine acetyltransferase from ara- bidopsis thaliana carries a bifunctional domain for catalysis and protein– protein interaction,”European Journal of Biochemistry, vol. 268, no. 3, pp. 686–693, 2001. ZHUet al.: STRUCTURE-GUIDED ADAPTIVE PROPAGATION FO...
2001
-
[4]
A mouse protein interactome through combined literature mining with multiple sources of interaction evidence,
X. Li, H. Cai, J. Xu, S. Ying, and Y . Zhang, “A mouse protein interactome through combined literature mining with multiple sources of interaction evidence,”Amino Acids, vol. 38, no. 4, pp. 1237–1252, 2010
2010
-
[5]
Review and comparative assessment of sequence-based predictors of protein-binding residues,
J. Zhang and L. Kurgan, “Review and comparative assessment of sequence-based predictors of protein-binding residues,”Briefings in bioinformatics, vol. 19, no. 5, pp. 821–837, 2018
2018
-
[6]
Edg- ppis: an equivariant and dual-scale graph network for protein–protein interaction site prediction,
Z. Zhang, Z. Li, W. Li, Q. Zhang, J. Xiao, S. Ding, and Y . Han, “Edg- ppis: an equivariant and dual-scale graph network for protein–protein interaction site prediction,”BMC genomics, vol. 26, no. 1, p. 862, 2025
2025
-
[7]
More challenges for machine-learning protein interactions,
T. Hamp and B. Rost, “More challenges for machine-learning protein interactions,”Bioinformatics, vol. 31, no. 10, pp. 1521–1525, 2015
2015
-
[8]
Deciphering protein–protein interactions. part i. experimental techniques and databases,
B. A. Shoemaker and A. R. Panchenko, “Deciphering protein–protein interactions. part i. experimental techniques and databases,”PLoS com- putational biology, vol. 3, no. 3, p. e42, 2007
2007
-
[9]
Applying the na ¨ıve bayes classifier with kernel density estimation to the prediction of protein–protein interaction sites,
Y . Murakami and K. Mizuguchi, “Applying the na ¨ıve bayes classifier with kernel density estimation to the prediction of protein–protein interaction sites,”Bioinformatics, vol. 26, no. 15, pp. 1841–1848, 2010
2010
-
[10]
Intpred: a structure-based predictor of protein–protein interaction sites,
T. C. Northey, A. Bare ˇsi´c, and A. C. Martin, “Intpred: a structure-based predictor of protein–protein interaction sites,”Bioinformatics, vol. 34, no. 2, pp. 223–229, 2018
2018
-
[11]
Scriber: accurate and partner type-specific prediction of protein-binding residues from proteins sequences,
J. Zhang and L. Kurgan, “Scriber: accurate and partner type-specific prediction of protein-binding residues from proteins sequences,”Bioin- formatics, vol. 35, no. 14, pp. i343–i353, 2019
2019
-
[12]
Convsppis: identifying protein-protein interaction sites by an ensemble convolutional neural network with feature graph,
H. Zhu, X. Du, and Y . Yao, “Convsppis: identifying protein-protein interaction sites by an ensemble convolutional neural network with feature graph,”Current Bioinformatics, vol. 15, no. 4, pp. 368–378, 2020
2020
-
[13]
Delphi: accurate deep ensemble model for protein interaction sites prediction,
Y . Li, G. B. Golding, and L. Ilie, “Delphi: accurate deep ensemble model for protein interaction sites prediction,”Bioinformatics, vol. 37, no. 7, pp. 896–904, 2021
2021
-
[14]
Structure-aware protein–protein interaction site prediction using deep graph convolu- tional network,
Q. Yuan, J. Chen, H. Zhao, Y . Zhou, and Y . Yang, “Structure-aware protein–protein interaction site prediction using deep graph convolu- tional network,”Bioinformatics, vol. 38, no. 1, pp. 125–132, 2022
2022
-
[15]
Asce-ppis: a protein–protein interaction sites predictor based on equivariant graph neural network with fusion of structure-aware pooling and graph collapse,
G. Shen, Z. Zhang, Z. Deng, X. Pan, H.-B. Shen, D.-J. Yu, S. Hu, and Y . Ge, “Asce-ppis: a protein–protein interaction sites predictor based on equivariant graph neural network with fusion of structure-aware pooling and graph collapse,”Bioinformatics, vol. 41, no. 8, p. btaf423, 2025
2025
-
[16]
Mgma-ppis: Predicting the protein–protein interaction site with multiview graph embedding and multiscale attention fusion,
Y . Han, S.-W. Zhang, Q.-Q. Zhang, and M.-H. Shi, “Mgma-ppis: Predicting the protein–protein interaction site with multiview graph embedding and multiscale attention fusion,”GigaScience, vol. 14, p. giaf114, 2025
2025
-
[17]
Comgat-ppis: A community-augmented graph attention network for protein-protein interaction site prediction,
Y . Zhang, Q. Bao, Y . Luo, Z. Li, M. Ma, E. Zhu, and J. Xu, “Comgat-ppis: A community-augmented graph attention network for protein-protein interaction site prediction,” in2025 IEEE International Conference on Bioinformatics and Biomedicine (BIBM). IEEE, 2025, pp. 459–462
2025
-
[18]
Conservation and relative importance of residues across protein-protein interfaces,
M. Guharoy and P. Chakrabarti, “Conservation and relative importance of residues across protein-protein interfaces,”Proceedings of the Na- tional Academy of Sciences, vol. 102, no. 43, pp. 15 447–15 452, 2005
2005
-
[19]
A simple definition of structural regions in proteins and its use in analyzing interface evolution,
E. D. Levy, “A simple definition of structural regions in proteins and its use in analyzing interface evolution,”Journal of molecular biology, vol. 403, no. 4, pp. 660–670, 2010
2010
-
[20]
The contribution of missense mutations in core and rim residues of protein–protein interfaces to human disease,
A. David and M. J. Sternberg, “The contribution of missense mutations in core and rim residues of protein–protein interfaces to human disease,” Journal of molecular biology, vol. 427, no. 17, pp. 2886–2898, 2015
2015
-
[21]
Local geometry and evolutionary con- servation of protein surfaces reveal the multiple recognition patches in protein-protein interactions,
E. Laine and A. Carbone, “Local geometry and evolutionary con- servation of protein surfaces reveal the multiple recognition patches in protein-protein interactions,”PLoS computational biology, vol. 11, no. 12, p. e1004580, 2015
2015
-
[22]
Predict then propagate: Graph neural networks meet personalized pagerank,
J. Gasteiger, A. Bojchevski, and S. G ¨unnemann, “Predict then propagate: Graph neural networks meet personalized pagerank,” inInternational Conference on Learning Representations, 2019
2019
-
[23]
Zeng, F.-F
X. Zeng, F.-F. Meng, X. Li, K.-Y . Zhong, B. Jiang, and Y . Li, “Ghgpr- ppis: a graph convolutional network for identifying protein-protein interaction site using heat kernel with generalized pagerank techniques and edge self-attention feature processing block,”Computers in Biology and Medicine, vol. 168, p. 107683, 2024
2024
-
[24]
Agat-ppis: a novel protein–protein interaction site predictor based on augmented graph attention network with initial residual and identity mapping,
Y . Zhou, Y . Jiang, and Y . Yang, “Agat-ppis: a novel protein–protein interaction site predictor based on augmented graph attention network with initial residual and identity mapping,”Briefings in bioinformatics, vol. 24, no. 3, p. bbad122, 2023
2023
-
[25]
Gapped blast and psi-blast: a new generation of protein database search programs,
S. F. Altschul, T. L. Madden, A. A. Sch ¨affer, J. Zhang, Z. Zhang, W. Miller, and D. J. Lipman, “Gapped blast and psi-blast: a new generation of protein database search programs,”Nucleic acids research, vol. 25, no. 17, pp. 3389–3402, 1997
1997
-
[26]
Hhblits: lightning- fast iterative protein sequence searching by hmm-hmm alignment,
M. Remmert, A. Biegert, A. Hauser, and J. S ¨oding, “Hhblits: lightning- fast iterative protein sequence searching by hmm-hmm alignment,” Nature methods, vol. 9, no. 2, pp. 173–175, 2012
2012
-
[27]
Dictionary of protein secondary struc- ture: pattern recognition of hydrogen-bonded and geometrical features,
W. Kabsch and C. Sander, “Dictionary of protein secondary struc- ture: pattern recognition of hydrogen-bonded and geometrical features,” Biopolymers: Original Research on Biomolecules, vol. 22, no. 12, pp. 2577–2637, 1983
1983
-
[28]
E (n) equivariant graph neural networks,
V . G. Satorras, E. Hoogeboom, and M. Welling, “E (n) equivariant graph neural networks,” inInternational conference on machine learning. PMLR, 2021, pp. 9323–9332
2021
-
[29]
Semi-supervised classification with graph convolutional networks,
T. N. Kipf and M. Welling, “Semi-supervised classification with graph convolutional networks,” inInternational Conference on Learning Rep- resentations, 2017
2017
-
[30]
Deeper insights into graph convolutional networks for semi-supervised learning,
Q. Li, Z. Han, and X.-M. Wu, “Deeper insights into graph convolutional networks for semi-supervised learning,” inProceedings of the AAAI conference on artificial intelligence, vol. 32, 2018
2018
-
[31]
The pagerank citation ranking: Bringing order to the web. technical report,
L. Page, “The pagerank citation ranking: Bringing order to the web. technical report,”Stanford Digital Library Technologies Project, 1998, 1998
1998
-
[32]
Muse: Parallel multi-scale attention for sequence to sequence learning,
G. Zhao, X. Sun, J. Xu, Z. Zhang, and L. Luo, “Muse: Parallel multi-scale attention for sequence to sequence learning,”arXiv preprint arXiv:1911.09483, 2019
-
[33]
Pay Less Attention with Lightweight and Dynamic Convolutions
F. Wu, A. Fan, A. Baevski, Y . N. Dauphin, and M. Auli, “Pay less attention with lightweight and dynamic convolutions,”arXiv preprint arXiv:1901.10430, 2019
work page internal anchor Pith review Pith/arXiv arXiv 1901
-
[34]
Focal loss for dense object detection,
T.-Y . Lin, P. Goyal, R. Girshick, K. He, and P. Doll ´ar, “Focal loss for dense object detection,” inProceedings of the IEEE International Conference on Computer Vision, 2017, pp. 2980–2988
2017
-
[35]
Agf-ppis: a protein–protein interaction site predictor based on an attention mechanism and graph convolutional networks,
X. Fu, Y . Yuan, H. Qiu, H. Suo, Y . Song, A. Li, Y . Zhang, C. Xiao, Y . Li, L. Douet al., “Agf-ppis: a protein–protein interaction site predictor based on an attention mechanism and graph convolutional networks,” Methods, vol. 222, pp. 142–151, 2024
2024
-
[36]
Prona2020 predicts protein–dna, protein–rna, and protein–protein binding proteins and residues from sequence,
J. Qiu, M. Bernhofer, M. Heinzinger, S. Kemper, T. Norambuena, F. Melo, and B. Rost, “Prona2020 predicts protein–dna, protein–rna, and protein–protein binding proteins and residues from sequence,”Journal of molecular biology, vol. 432, no. 7, pp. 2428–2443, 2020
2020
-
[37]
Sequence-based prediction of protein-protein interaction sites by simplified long short- term memory network,
B. Zhang, J. Li, L. Quan, Y . Chen, and Q. L ¨u, “Sequence-based prediction of protein-protein interaction sites by simplified long short- term memory network,”Neurocomputing, vol. 357, pp. 86–100, 2019
2019
-
[38]
Protein– protein interaction site prediction through combining local and global features with deep neural networks,
M. Zeng, F. Zhang, F.-X. Wu, Y . Li, J. Wang, and M. Li, “Protein– protein interaction site prediction through combining local and global features with deep neural networks,”Bioinformatics, vol. 36, no. 4, pp. 1114–1120, 2020
2020
-
[39]
Prediction-based fingerprints of protein– protein interactions,
A. Porollo and J. Meller, “Prediction-based fingerprints of protein– protein interactions,”Proteins: Structure, Function, and Bioinformatics, vol. 66, no. 3, pp. 630–645, 2007
2007
-
[40]
Deciphering interaction fingerprints from protein molecular surfaces using geometric deep learning,
P. Gainza, F. Sverrisson, F. Monti, E. Rodola, D. Boscaini, M. M. Bronstein, and B. E. Correia, “Deciphering interaction fingerprints from protein molecular surfaces using geometric deep learning,”Nature methods, vol. 17, no. 2, pp. 184–192, 2020
2020
-
[41]
Deepprosite: structure-aware protein binding site prediction using esmfold and pretrained language model,
Y . Fang, Y . Jiang, L. Wei, Q. Ma, Z. Ren, Q. Yuan, and D.-Q. Wei, “Deepprosite: structure-aware protein binding site prediction using esmfold and pretrained language model,”Bioinformatics, vol. 39, no. 12, p. btad718, 2023
2023
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.