Deciphering Fingerprints of 3D Molecular Surfaces for Accurate Epitope Prediction
Pith reviewed 2026-06-26 09:05 UTC · model grok-4.3
The pith
SurfBind predicts epitopes by processing 3D molecular surfaces directly with a Transformer that uses patch modeling and binder-aware cross-attention.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
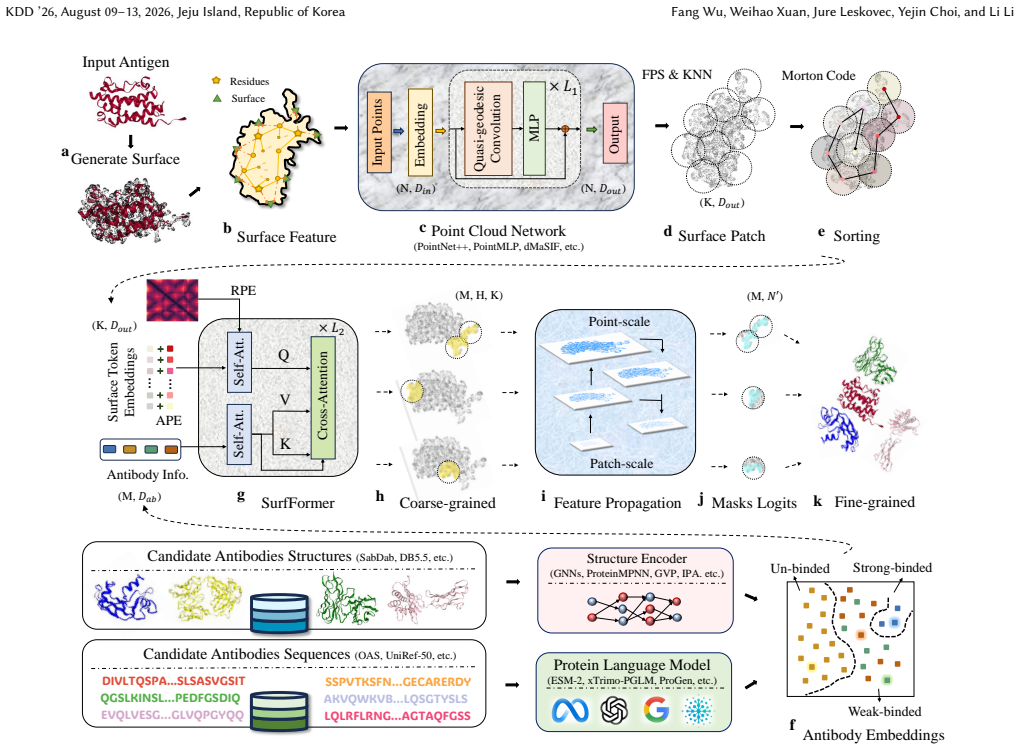
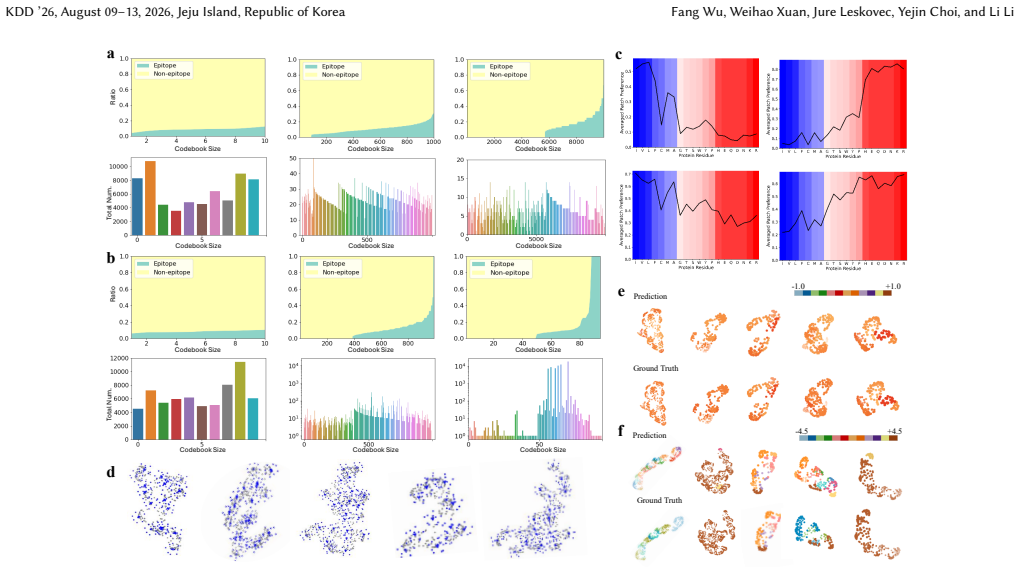
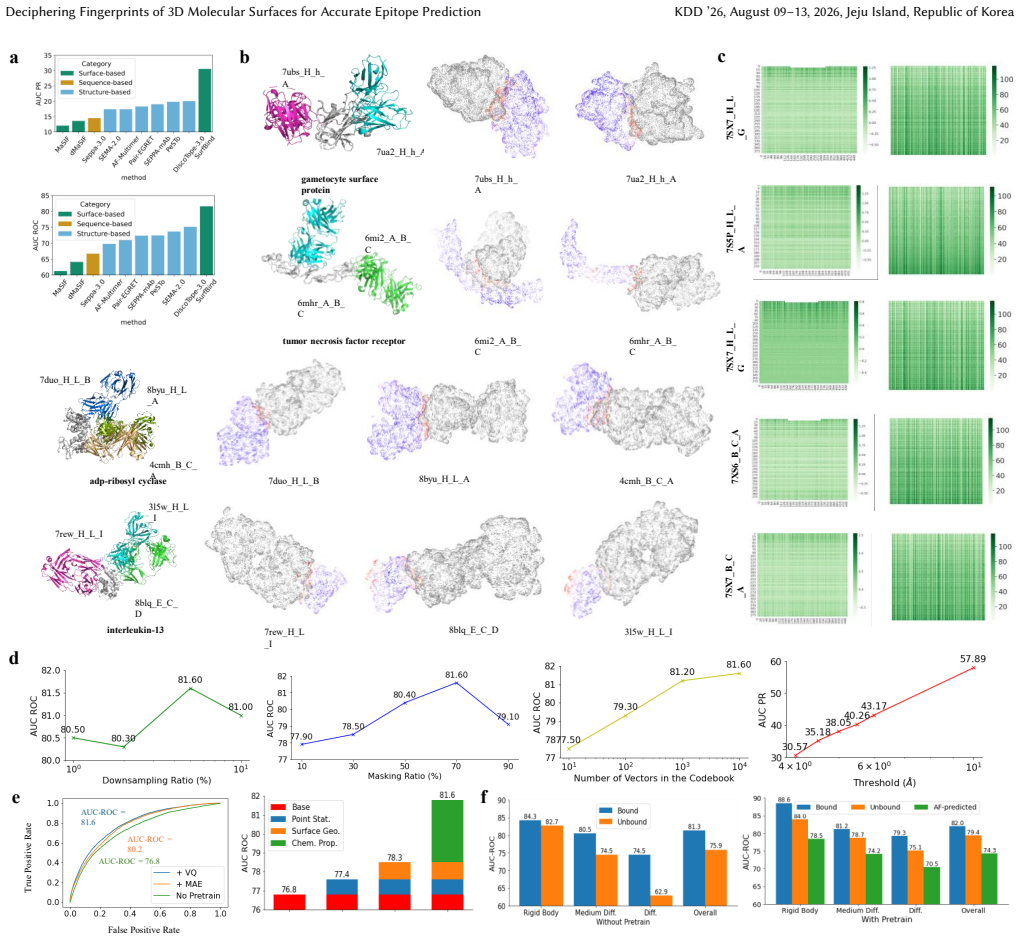
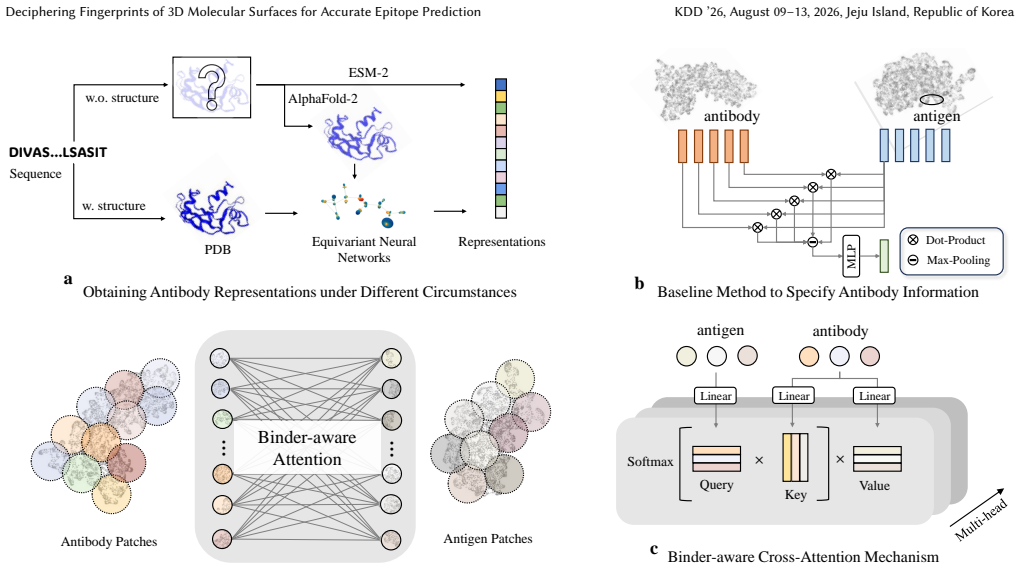
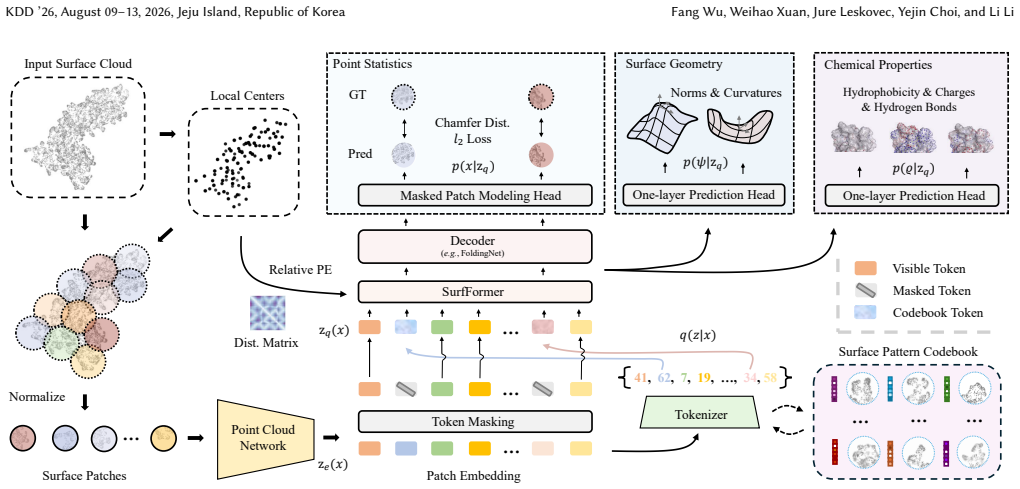
SurfBind is a surface-centric learning framework for epitope prediction that operates directly on molecular surface representations. It integrates geometric and physicochemical cues through a Transformer-based architecture with patch-level surface modeling, binder-aware cross-attention, and a hierarchical coarse-to-fine prediction paradigm. Experiments on challenging epitope identification benchmarks, including SAbDab and DB5.5, demonstrate that SurfBind achieves state-of-the-art performance and strong generalization across unseen antibodies and conformational states.
What carries the argument
The binder-aware cross-attention mechanism within a Transformer that processes patch-level molecular surface representations to integrate geometric and physicochemical cues.
If this is right
- Improved epitope prediction enables more precise design of antibody-based therapies.
- The framework shows that surface geometry matters more than linear sequence information for identifying binding sites.
- Strong generalization indicates the method can handle novel protein complexes in drug discovery.
- Hierarchical prediction allows efficient processing of complex 3D molecular structures.
Where Pith is reading between the lines
- Surface modeling approaches may extend to predicting other molecular interactions such as enzyme-substrate binding.
- Integration with experimental data like cryo-EM could be tested to further refine predictions.
- Similar surface-centric models could advance related tasks such as protein docking.
Load-bearing premise
Molecular surface representations processed through the Transformer with patch-level modeling and binder-aware cross-attention capture the geometric and physicochemical patterns that determine antibody-antigen recognition more effectively than sequence or backbone alternatives.
What would settle it
A new benchmark with diverse conformational states where SurfBind fails to outperform sequence-based methods on epitope identification would show that the surface approach does not capture the determining patterns.
Figures

read the original abstract
Molecular surfaces encode the geometric and physicochemical patterns that determine antibody-antigen recognition, central to epitope prediction. However, existing methods rely on sequences or backbone structures and struggle to capture discontinuous, surface-driven epitopes. This study presents SurfBind, a surface-centric learning framework for epitope prediction that operates directly on molecular surface representations. SurfBind integrates geometric and physicochemical cues through a Transformer-based architecture with patch-level surface modeling, binder-aware cross-attention, and a hierarchical coarse-to-fine prediction paradigm. Experiments on challenging epitope identification benchmarks, including SAbDab and DB5.5, demonstrate that SurfBind achieves state-of-the-art performance and strong generalization across unseen antibodies and conformational states, highlighting the value of interaction-aware surface modeling for understanding the crucial mechanisms of protein-protein interactions.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript introduces SurfBind, a surface-centric framework for epitope prediction that processes 3D molecular surface representations using a Transformer architecture incorporating patch-level modeling, binder-aware cross-attention, and a hierarchical coarse-to-fine prediction paradigm. It claims state-of-the-art performance and strong generalization across unseen antibodies and conformational states on benchmarks including SAbDab and DB5.5, arguing that surface representations better capture discontinuous epitopes than sequence or backbone-based methods.
Significance. If the empirical claims hold after proper validation, the work would provide evidence that direct modeling of molecular surfaces can improve epitope identification by capturing geometric and physicochemical interaction patterns, potentially shifting the field from sequence/backbone-centric approaches toward surface-aware methods for protein-protein interaction analysis.
major comments (2)
- [Abstract] Abstract: The central claim that SurfBind 'achieves state-of-the-art performance' on SAbDab and DB5.5 is unsupported because the text supplies no quantitative results, baselines, error bars, statistical tests, or performance numbers, rendering the claim unevaluable.
- [Experiments] Experiments section (implied by benchmark references): No details are provided on data splits, training procedures, evaluation protocols, ablation studies, or how generalization to unseen antibodies was ensured, which directly undermines assessment of whether reported improvements are independent of fitting choices or data selection.
Simulated Author's Rebuttal
We thank the referee for the constructive feedback on our manuscript. We address each major comment below and are prepared to revise the manuscript accordingly to improve clarity and support for our claims.
read point-by-point responses
-
Referee: [Abstract] Abstract: The central claim that SurfBind 'achieves state-of-the-art performance' on SAbDab and DB5.5 is unsupported because the text supplies no quantitative results, baselines, error bars, statistical tests, or performance numbers, rendering the claim unevaluable.
Authors: We agree that the abstract does not contain specific quantitative results, baselines, or statistical details. While the full manuscript's Experiments section reports these comparisons (including metrics on SAbDab and DB5.5), we will revise the abstract to incorporate key performance numbers and baseline references to make the state-of-the-art claim directly supported within the abstract itself. revision: yes
-
Referee: [Experiments] Experiments section (implied by benchmark references): No details are provided on data splits, training procedures, evaluation protocols, ablation studies, or how generalization to unseen antibodies was ensured, which directly undermines assessment of whether reported improvements are independent of fitting choices or data selection.
Authors: The manuscript references the benchmarks and generalization to unseen antibodies, but we acknowledge that explicit details on data splits, training procedures, evaluation protocols, and ablation studies could be expanded for full transparency. We will revise the Experiments section to include these specifics, such as definitions of unseen antibody splits and ablation results, to allow independent assessment of the improvements. revision: yes
Circularity Check
No significant circularity detected
full rationale
The paper describes an empirical machine-learning framework (SurfBind) whose central claims rest on reported performance numbers on external public benchmarks (SAbDab, DB5.5). No derivation chain, equations, or self-citation load-bearing steps are present in the supplied text that would reduce a claimed result to a fitted input or self-definition by construction. The architecture choices are presented as design decisions whose value is asserted via experiment, not via internal tautology. This is the normal case for an applied ML paper whose validity is externally falsifiable on held-out data.
Axiom & Free-Parameter Ledger
free parameters (2)
- Transformer weights and attention parameters
- Patch definition and hierarchical prediction thresholds
axioms (1)
- domain assumption Molecular surfaces encode the geometric and physicochemical patterns that determine antibody-antigen recognition
Reference graph
Works this paper leans on
-
[1]
Ramisa Alam, Sazan Mahbub, and Md Shamsuzzoha Bayzid. 2023. Pair-EGRET: enhancing the prediction of protein-protein interaction sites through graph attention networks and protein language models.bioRxiv(2023), 2023–12
2023
-
[2]
Hifzur Rahman Ansari and Gajendra PS Raghava. 2010. Identification of confor- mational B-cell Epitopes in an antigen from its primary sequence.Immunome research6, 1 (2010), 1–9
2010
-
[3]
Sankar Basu and Björn Wallner. 2016. DockQ: a quality measure for protein- protein docking models.PloS one11, 8 (2016), e0161879
2016
-
[4]
SYED NISAR HUSSAIN BUKHARI, MUNEER AHMAD DAR, and MUJTABA SHAFI. 2021. USING RANDOM FOREST TO PREDICT T-CELL EPITOPES OF DENGUE VIRUS. (2021)
2021
-
[5]
Syed Nisar Hussain Bukhari, Amit Jain, Ehtishamul Haq, Abolfazl Mehbodniya, and Julian Webber. 2022. Machine learning techniques for the prediction of B-cell and T-cell epitopes as potential vaccine targets with a specific focus on SARS-CoV-2 pathogen: A review.Pathogens11, 2 (2022), 146
2022
-
[6]
Guangyan Chen, Meiling Wang, Yi Yang, Kai Yu, Li Yuan, and Yufeng Yue. 2023. PointGPT: Auto-regressively Generative Pre-training from Point Clouds.arXiv preprint arXiv:2305.11487(2023)
arXiv 2023
-
[7]
Junwei Chen, Bowen Zhao, Shenggeng Lin, Heqi Sun, Xueying Mao, Meng Wang, Yanyi Chu, Liang Hong, Dong-Qing Wei, Min Li, et al. 2024. TEPCAM: Prediction of T-cell receptor–epitope binding specificity via interpretable deep learning. Protein Science33, 1 (2024), e4841
2024
-
[8]
Gabriel Cia, Fabrizio Pucci, and Marianne Rooman. 2023. Critical review of conformational B-cell epitope prediction methods.Briefings in bioinformatics24, 1 (2023), bbac567
2023
-
[9]
Joakim Noddeskov Clifford, Magnus Haraldson Hoie, Sebastian Deleuran, Bjoern Peters, Morten Nielsen, and Paolo Marcatili. 2022. BepiPred-3.0: Improved B-cell epitope prediction using protein language models.Protein Science31, 12 (2022), e4497
2022
-
[10]
Bruna Moreira da Silva, YooChan Myung, David B Ascher, and Douglas EV Pires
-
[11]
epitope3D: a machine learning method for conformational B-cell epitope prediction.Briefings in Bioinformatics23, 1 (2022), bbab423
2022
-
[12]
Alice Del Vecchio, Andreea Deac, Pietro Liò, and Petar Veličković. 2021. Neu- ral message passing for joint paratope-epitope prediction.arXiv preprint arXiv:2106.00757(2021)
arXiv 2021
-
[13]
Arthur Deng, Karsten Householder, Fang Wu, Sebastian Thrun, K Christopher Garcia, and Brian Trippe. 2025. Predicting mutational effects on protein binding from folding energy.arXiv preprint arXiv:2507.05502(2025)
arXiv 2025
-
[14]
Bin Deng, Shaolong Zhu, Andrew M Macklin, Jianrong Xu, Cristina Lento, Adnan Sljoka, and Derek J Wilson. 2017. Suppressing allostery in epitope mapping experiments using millisecond hydrogen/deuterium exchange mass spectrometry. InMAbs, Vol. 9. Taylor & Francis, 1327–1336
2017
-
[15]
Dattatraya V Desai and Urmila Kulkarni-Kale. 2014. T-cell epitope prediction methods: an overview.Immunoinformatics(2014), 333–364
2014
-
[16]
Tom Duff, James Burgess, Per Christensen, Christophe Hery, Andrew Kensler, Max Liani, and Ryusuke Villemin. 2017. Building an orthonormal basis, revisited. JCGT6, 1 (2017)
2017
-
[17]
James Dunbar, Konrad Krawczyk, Jinwoo Leem, Terry Baker, Angelika Fuchs, Guy Georges, Jiye Shi, and Charlotte M Deane. 2014. SAbDab: the structural antibody database.Nucleic acids research42, D1 (2014), D1140–D1146
2014
-
[18]
Yasser EL-Manzalawy, Drena Dobbs, and Vasant Honavar. 2008. Predicting linear B-cell epitopes using string kernels.Journal of Molecular Recognition: An Interdisciplinary Journal21, 4 (2008), 243–255
2008
-
[19]
Reyhaneh Esmaielbeiki, Konrad Krawczyk, Bernhard Knapp, Jean-Christophe Nebel, and Charlotte M Deane. 2016. Progress and challenges in predicting protein interfaces.Briefings in bioinformatics17, 1 (2016), 117–131
2016
-
[20]
Richard Evans, Michael O’Neill, Alexander Pritzel, Natasha Antropova, Andrew Senior, Tim Green, Augustin Zidek, Russ Bates, Sam Blackwell, Jason Yim, et al
-
[21]
Protein complex prediction with AlphaFold-Multimer.biorxiv(2021), 2021–10
2021
-
[22]
Haoqiang Fan, Hao Su, and Leonidas J Guibas. 2017. A point set generation network for 3d object reconstruction from a single image. InProceedings of the IEEE conference on computer vision and pattern recognition. 605–613
2017
-
[23]
Pablo Gainza, Freyr Sverrisson, Frederico Monti, Emanuele Rodola, D Boscaini, MM Bronstein, and BE Correia. 2020. Deciphering interaction fingerprints from protein molecular surfaces using geometric deep learning.Nature Methods17, 2 (2020), 184–192
2020
-
[24]
Kaiming He, Xinlei Chen, Saining Xie, Yanghao Li, Piotr Dollar, and Ross Girshick
-
[25]
InProceedings of the IEEE/CVF conference on computer vision and pattern recognition
Masked autoencoders are scalable vision learners. InProceedings of the IEEE/CVF conference on computer vision and pattern recognition. 16000–16009
-
[26]
Magnus Haraldson Hoie, Frederik Steensgaard Gade, Julie Maria Johansen, Char- lotte Wurtzen, Ole Winther, Morten Nielsen, and Paolo Marcatili. 2024. DiscoTope- 3.0: improved B-cell epitope prediction using inverse folding latent representa- tions.Frontiers in Immunology15 (2024), 1322712
2024
-
[27]
Chloe Hsu, Robert Verkuil, Jason Liu, Zeming Lin, Brian Hie, Tom Sercu, Adam Lerer, and Alexander Rives. 2022. Learning inverse folding from millions of predicted structures.bioRxiv(2022)
2022
-
[28]
Eric Jang, Shixiang Gu, and Ben Poole. 2016. Categorical reparameterization with gumbel-softmax.arXiv preprint arXiv:1611.01144(2016)
Pith/arXiv arXiv 2016
-
[29]
Martin Closter Jespersen, Bjoern Peters, Morten Nielsen, and Paolo Marcatili
-
[30]
BepiPred-2.0: improving sequence-based B-cell epitope prediction using conformational epitopes.Nucleic acids research45, W1 (2017), W24–W29
2017
-
[31]
Yize Jiang, Xinze Li, Yuanyuan Zhang, Jin Han, Youjun Xu, Ayush Pandit, Zaixi Zhang, Mengdi Wang, Mengyang Wang, Minjie Shen, et al . 2025. PoseX: AI Defeats Physics Approaches on Protein-Ligand Cross Docking.arXiv preprint arXiv:2505.01700(2025)
Pith/arXiv arXiv 2025
-
[32]
Wengong Jin, Regina Barzilay, and Tommi Jaakkola. 2022. Antibody-Antigen Docking and Design via Hierarchical Equivariant Refinement.arXiv preprint arXiv:2207.06616(2022)
arXiv 2022
-
[33]
Robbie P Joosten, Fei Long, Garib N Murshudov, and Anastassis Perrakis. 2014. The PDB_REDO server for macromolecular structure model optimization.IUCrJ 1, 4 (2014), 213–220
2014
-
[34]
Diederik P Kingma and Jimmy Ba. 2014. Adam: A method for stochastic opti- mization.arXiv preprint arXiv:1412.6980(2014)
Pith/arXiv arXiv 2014
-
[35]
Lucien F Krapp, Luciano A Abriata, Fabio Cortes Rodriguez, and Matteo Dal Per- aro. 2023. PeSTo: parameter-free geometric deep learning for accurate prediction of protein binding interfaces.Nature Communications14, 1 (2023), 2175
2023
-
[36]
Jinwoo Leem, Laura S Mitchell, James HR Farmery, Justin Barton, and Jacob D Galson. 2022. Deciphering the language of antibodies using self-supervised learning.Patterns(2022), 100513
2022
-
[37]
Guanlue Li, Xufeng Zhao, Fang Wu, and Sören Laue. 2026. Joint design of protein surface and backbone using a diffusion bridge model.Advances in Neural Information Processing Systems38 (2026), 169682–169708
2026
-
[38]
Pengpai Li and Zhi-Ping Liu. 2023. GeoBind: segmentation of nucleic acid binding interface on protein surface with geometric deep learning.Nucleic Acids Research 51, 10 (2023), e60–e60
2023
-
[39]
Siyuan Li, Luyuan Zhang, Zedong Wang, Di Wu, Lirong Wu, Zicheng Liu, Jun Xia, Cheng Tan, Yang Liu, Baigui Sun, et al. 2023. Masked Modeling for Self-supervised Representation Learning on Vision and Beyond.arXiv preprint arXiv:2401.00897 (2023)
arXiv 2023
-
[40]
Tsung-Yi Lin, Priya Goyal, Ross Girshick, Kaiming He, and Piotr Dollar. 2017. Focal loss for dense object detection. InProceedings of the IEEE international conference on computer vision. 2980–2988
2017
-
[41]
Zeming Lin et al. 2022. Language models of protein sequences at the scale of evolution enable accurate structure prediction.bioRxiv(2022)
2022
-
[42]
ChuNan Liu, Lilian Denzler, Yihong Chen, Andrew Martin, and Brooks Paige
-
[43]
AsEP: Benchmarking deep learning methods for antibody-specific epitope prediction.Advances in Neural Information Processing Systems37 (2024), 11700– 11734
2024
-
[44]
Zhihai Liu, Yan Li, Li Han, Jie Li, Jie Liu, Zhixiong Zhao, Wei Nie, Yuchen Liu, and Renxiao Wang. 2015. PDB-wide collection of binding data: current status of the PDBbind database.Bioinformatics31, 3 (2015), 405–412
2015
-
[45]
Shitong Luo, Yufeng Su, Zuofan Wu, Chenpeng Su, Jian Peng, and Jianzhu Ma
-
[46]
Rotamer Density Estimator is an Unsupervised Learner of the Effect of Mutations on Protein-Protein Interaction.bioRxiv(2023), 2023–02
2023
-
[47]
Matt McPartlon and Jinbo Xu. 2023. Deep learning for flexible and site-specific protein docking and design.BioRxiv(2023), 2023–04
2023
-
[48]
Niloy J Mitra and An Nguyen. 2003. Estimating surface normals in noisy point cloud data. InProceedings of the nineteenth annual symposium on Computational geometry. 322–328
2003
-
[49]
Guy M Morton. 1966. A computer oriented geodetic data base and a new technique in file sequencing. (1966)
1966
-
[50]
Stelios K Mylonas, Apostolos Axenopoulos, and Petros Daras. 2021. DeepSurf: a surface-based deep learning approach for the prediction of ligand binding sites on proteins.Bioinformatics37, 12 (2021), 1681–1690
2021
-
[51]
Bjoern Peters, Morten Nielsen, and Alessandro Sette. 2020. T cell epitope predic- tions.Annual Review of Immunology38, 1 (2020), 123–145
2020
-
[52]
Ksenia Polonsky, Tal Pupko, and Natalia T Freund. 2023. Evaluation of the Ability of AlphaFold to Predict the Three-Dimensional Structures of Antibodies and KDD ’26, August 09–13, 2026, Jeju Island, Republic of Korea Fang Wu, Weihao Xuan, Jure Leskovec, Yejin Choi, and Li Li Epitopes.The Journal of Immunology211, 10 (2023), 1578–1588
2023
-
[53]
Lenka Potocnakova, Mangesh Bhide, and Lucia Borszekova Pulzova. 2016. An introduction to B-cell epitope mapping and in silico epitope prediction.Journal of immunology research2016, 1 (2016), 6760830
2016
-
[54]
Charles Ruizhongtai Qi, Li Yi, Hao Su, and Leonidas J Guibas. 2017. Pointnet++: Deep hierarchical feature learning on point sets in a metric space.Advances in neural information processing systems30 (2017)
2017
-
[55]
Huan Qi, Mingliang Ma, Chuansheng Hu, Zhao-wei Xu, Fan-lin Wu, Nan Wang, Dan-yun Lai, Yang Li, Hainan Zhang, He-wei Jiang, et al. 2021. Antibody binding epitope mapping (AbMap) of hundred antibodies in a single run.Molecular & Cellular Proteomics20 (2021)
2021
-
[56]
Tianyi Qiu, Lu Zhang, Zikun Chen, Yuan Wang, Tiantian Mao, Caicui Wang, Yewei Cun, Genhui Zheng, Deyu Yan, Mengdi Zhou, et al. 2023. SEPPA-mAb: spatial epitope prediction of protein antigens for mAbs.Nucleic Acids Research (2023), gkad427
2023
-
[57]
Aditya Ramesh, Mikhail Pavlov, Gabriel Goh, Scott Gray, Chelsea Voss, Alec Radford, Mark Chen, and Ilya Sutskever. 2021. Zero-shot text-to-image generation. InInternational Conference on Machine Learning. PMLR, 8821–8831
2021
-
[58]
Saleh Riahi, Jae Hyeon Lee, Taylor Sorenson, Shuai Wei, Sven Jager, Reza Olfati- Saber, Yanfeng Zhou, Anna Park, Maria Wendt, Herve Minoux, et al. 2023. Surface ID: a geometry-aware system for protein molecular surface comparison.Bioin- formatics39, 4 (2023), btad196
2023
-
[59]
Alexander Rives et al . 2021. Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences.Proceedings of the National Academy of Sciences118, 15 (2021), e2016239118
2021
-
[60]
Jose L Sanchez-Trincado, Marta Gomez-Perosanz, and Pedro A Reche. 2017. Fundamentals and methods for T-and B-cell epitope prediction.Journal of im- munology research2017, 1 (2017), 2680160
2017
-
[61]
Kristof T Schutt, Huziel E Sauceda, P-J Kindermans, Alexandre Tkatchenko, and K-R Muller. 2018. Schnet–a deep learning architecture for molecules and materials.The Journal of Chemical Physics148, 24 (2018)
2018
-
[62]
Tatiana I Shashkova et al. 2022. SEMA: Antigen B-cell conformational epitope prediction using deep transfer learning.Frontiers in immunology(2022), 5272
2022
-
[63]
Ruth E Soria-Guerra, Ricardo Nieto-Gomez, Dania O Govea-Alonso, and Sergio Rosales-Mendoza. 2015. An overview of bioinformatics tools for epitope predic- tion: implications on vaccine development.Journal of biomedical informatics53 (2015), 405–414
2015
-
[64]
Vitalii Stebliankin, Azam Shirali, Prabin Baral, Jimeng Shi, Prem Chapagain, Kalai Mathee, and Giri Narasimhan. 2023. Evaluating protein binding interfaces with transformer networks.Nature Machine Intelligence5, 9 (2023), 1042–1053
2023
-
[65]
Martin Steinegger and Johannes Soding. 2017. MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets.Nature biotechnology 35, 11 (2017), 1026–1028
2017
-
[66]
Freyr Sverrisson, Jean Feydy, Bruno E Correia, and Michael M Bronstein. 2021. Fast end-to-end learning on protein surfaces. InProceedings of the IEEE/CVF Conference on Computer Vision and Pattern Recognition. 15272–15281
2021
-
[67]
Xiaoyu Tian, Haoxi Ran, Yue Wang, and Hang Zhao. 2023. GeoMAE: Masked Geometric Target Prediction for Self-supervised Point Cloud Pre-Training. In Proceedings of the IEEE/CVF Conference on Computer Vision and Pattern Recognition. 13570–13580
2023
-
[68]
Jerome Tubiana, Dina Schneidman-Duhovny, and Haim J Wolfson. 2022. Scan- Net: an interpretable geometric deep learning model for structure-based protein binding site prediction.Nature Methods19, 6 (2022), 730–739
2022
-
[69]
Aaron Van Den Oord, Oriol Vinyals, et al. 2017. Neural discrete representation learning.Advances in neural information processing systems30 (2017)
2017
-
[70]
Mihaly Varadi, Stephen Anyango, Mandar Deshpande, Sreenath Nair, Cindy Natassia, Galabina Yordanova, David Yuan, Oana Stroe, Gemma Wood, Agata Laydon, et al. 2022. AlphaFold Protein Structure Database: massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic acids research50, D1 (2022), D439–D444
2022
-
[71]
Mihaly Varadi, Damian Bertoni, Paulyna Magana, Urmila Paramval, Ivanna Pidruchna, Malarvizhi Radhakrishnan, Maxim Tsenkov, Sreenath Nair, Milot Mirdita, Jingi Yeo, et al. 2024. AlphaFold Protein Structure Database in 2024: providing structure coverage for over 214 million protein sequences.Nucleic Acids Research52, D1 (2024), D368–D375
2024
-
[72]
Ashish Vaswani, Noam Shazeer, Niki Parmar, Jakob Uszkoreit, Llion Jones, Aidan N Gomez, Lukasz Kaiser, and Illia Polosukhin. 2017. Attention is all you need.Advances in neural information processing systems30 (2017)
2017
-
[73]
Thom Vreven et al. 2015. Updates to the integrated protein–protein interaction benchmarks: docking benchmark version 5 and affinity benchmark version 2. Journal of molecular biology427, 19 (2015), 3031–3041
2015
-
[74]
Xiao Wang, Genki Terashi, Charles W Christoffer, Mengmeng Zhu, and Daisuke Kihara. 2020. Protein docking model evaluation by 3D deep convolutional neural networks.Bioinformatics36, 7 (2020), 2113–2118
2020
-
[75]
Fang Wu. 2024. A semi-supervised molecular learning framework for activity cliff estimation. InProceedings of the Thirty-Third International Joint Conference on Artificial Intelligence. 6080–6088
2024
-
[76]
Fang Wu. 2025. DiffAntiSeq: A Controllable Diffusion Model for Efficient Anti- body Library Design. InLLM for Scientific Discovery: Reasoning, Assistance, and Collaboration
2025
-
[77]
Fang Wu. 2026. A semi-supervised molecular learning framework for activity cliff estimation.arXiv preprint arXiv:2601.04507(2026)
arXiv 2026
-
[78]
Fang Wu, Shuting Jin, Yinghui Jiang, Xurui Jin, Bowen Tang, Zhangming Niu, Xiangrong Liu, Qiang Zhang, Xiangxiang Zeng, and Stan Z Li. 2022. Pre-Training of Equivariant Graph Matching Networks with Conformation Flexibility for Drug Binding.Advanced Science9, 33 (2022), 2203796
2022
-
[79]
Fang Wu, Shuting Jin, Xiangru Tang, Junlin Xu, Mark Gerstein, Li Erran Li, and James Zou. 2026. D-flow: Multi-modality flow matching for d-peptide design. IEEE Journal of Biomedical and Health Informatics(2026)
2026
-
[80]
Fang Wu, Siyuan Li, Lirong Wu, Stan Z Li, Dragomir Radev, and Qiang Zhang
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.