ML-MAWS: Alignment-Free Maximum Likelihood Phylogeny Estimation Using Minimal Absent Words
Pith reviewed 2026-06-25 19:28 UTC · model grok-4.3
The pith
Encoding minimal absent words as binary characters enables maximum likelihood tree estimation on unaligned genomes.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
ML-MAWS recovers near-correct splits on bacterial, mitochondrial, viral and simulated genome benchmarks by encoding minimal absent words as a binary character matrix and estimating trees under the Lewis Mkv model with ascertainment bias correction; the method supplies per-branch statistical support and a probabilistic framework that distance-based alignment-free approaches lack.
What carries the argument
Binary presence/absence matrix of minimal absent words, filtered by strand awareness, entropy selection across lengths, and parsimony-informative capping, then analysed under the Lewis Mkv model with ascertainment bias correction.
If this is right
- Trees produced by ML-MAWS carry per-branch statistical support values unavailable from distance-only alignment-free methods.
- The method supplies a full probabilistic model that can be extended to model testing or ancestral-state reconstruction.
- Computation remains linear in sequence length because minimal absent words are extracted via suffix-automaton traversal.
- The same binary matrix can be re-analysed under alternative substitution models once the Lewis Mkv baseline is established.
Where Pith is reading between the lines
- Because the input is a character matrix rather than pairwise distances, the framework could be combined with existing coalescent or multispecies models that operate on character data.
- The entropy-based length selection step might generalise to other k-mer or word-based features beyond minimal absent words.
- If the binary signal proves robust, the approach could be applied to very large metagenomic assemblies where alignment is impractical.
Load-bearing premise
The binary presence or absence of minimal absent words still carries enough phylogenetic signal to produce trees whose topological accuracy matches or exceeds that of continuous distance methods after the described filtering.
What would settle it
A new benchmark collection in which the Robinson-Foulds or matching-split distance of ML-MAWS trees to the reference is substantially larger than that of published continuous distance baselines on the same sequences.
Figures

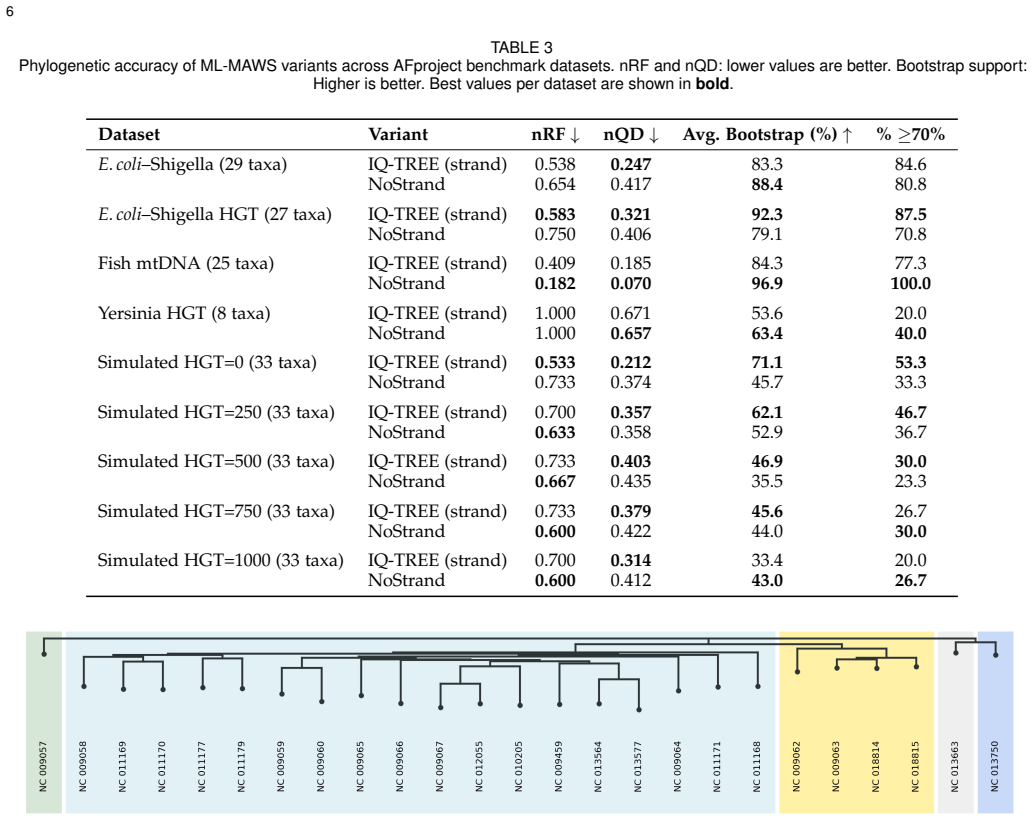
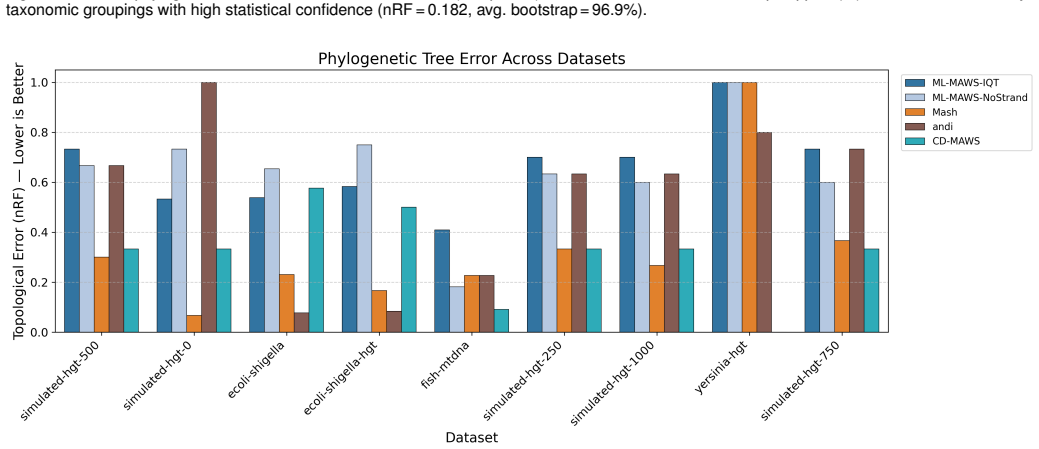



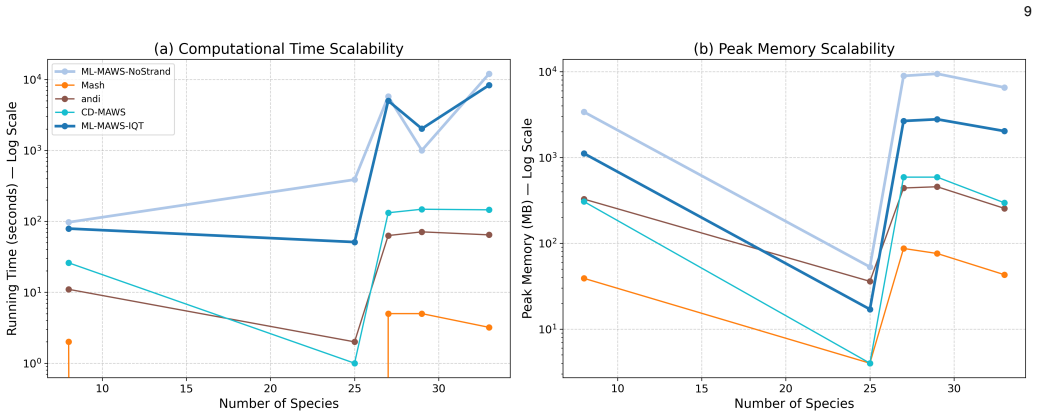
read the original abstract
Alignment-free methods in phylogenetic tree construction have major benefits in computational efficiency over alignment-based methods, but most sacrifice sequence information to pairwise distances, losing the statistical power of maximum likelihood (ML) inference. We describe ML-MAWS, an algorithm that fills this gap by encoding Minimal Absent Words (MAWs) as a binary presence/absence character matrix and estimating using an ML tree under the Lewis Mkv model using ascertainment bias correction. MAWs are obtained in linear time through the traversal of a suffix automaton. Three new elements contribute to the phylogenetic signal: strand-aware filtering combines forward and reverse complement MAW sets to eliminate compositional artifacts; entropy-based multi-length selection uses Shannon entropy maximization to select the most informative lengths of MAWs; and parsimony-informative character capping only retains the most discriminative columns. We tested ML-MAWS on 14 benchmark datasets of bacterial, mitochondrial, viral, and simulated genomes with normalized Robinson Foulds distances and matching split distances, against published reference trees. The results show that the coarse binary encoding of MAWs can lead to higher topological errors than continuous-valued distance baselines, while ML-MAWS can successfully recover near-correct splits and can uniquely provide per-branch statistical confidence as well as a rigorous probabilistic framework that is lacking in these methods.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript proposes ML-MAWS, an alignment-free phylogenetic method that extracts minimal absent words (MAWs) in linear time via suffix automata, encodes them as a binary presence/absence character matrix after strand-aware filtering, entropy-based length selection, and parsimony capping, and infers trees by maximum likelihood under the Lewis Mkv model with ascertainment bias correction. On 14 bacterial, mitochondrial, viral, and simulated datasets, it reports higher normalized Robinson-Foulds and matching-split distances than continuous distance baselines yet claims recovery of near-correct splits together with per-branch statistical supports and a probabilistic framework unavailable to distance methods.
Significance. If the central modeling assumptions hold, the work would supply the first explicit maximum-likelihood treatment of alignment-free data, a genuine methodological advance over purely distance-based approaches. The linear-time MAW extraction and the three filtering heuristics are computationally attractive and directly address known compositional artifacts. The explicit acknowledgment that topological accuracy remains inferior to distance baselines is a strength of the presentation, as it correctly frames the contribution around the availability of calibrated supports rather than raw accuracy.
major comments (2)
- [Abstract] Abstract: the claim that ML-MAWS 'can successfully recover near-correct splits' while simultaneously reporting higher topological error than distance baselines is load-bearing for the central contribution; the manuscript must define a quantitative threshold (e.g., normalized RF < 0.05 or matching-split distance < 0.10) and report per-dataset values to substantiate 'near-correct' rather than leaving the interpretation to the reader.
- [Methods] Methods (Lewis Mkv application and filtering pipeline): the model treats MAW columns as conditionally independent given the tree and branch lengths, yet each MAW is a deterministic function of the same input string; a single substitution can create or destroy multiple MAWs. The strand-aware union, entropy maximization, and parsimony capping steps are described but no diagnostic (e.g., pairwise character correlation matrix or simulation under a sequence evolution model) is provided to show that residual dependence is negligible. Because the per-branch supports and likelihood comparisons rest on this independence assumption, its violation directly undermines the claimed 'rigorous probabilistic framework'.
minor comments (2)
- The 14 benchmark datasets are referenced only generically; explicit accession numbers, sequence lengths, and reference tree sources should be tabulated to permit exact reproduction.
- Figure legends should state the exact normalization used for Robinson-Foulds distances and whether the reported values are means or medians across replicates.
Simulated Author's Rebuttal
We thank the referee for the constructive and detailed report. We address each major comment below and commit to revisions that directly respond to the concerns raised.
read point-by-point responses
-
Referee: [Abstract] Abstract: the claim that ML-MAWS 'can successfully recover near-correct splits' while simultaneously reporting higher topological error than distance baselines is load-bearing for the central contribution; the manuscript must define a quantitative threshold (e.g., normalized RF < 0.05 or matching-split distance < 0.10) and report per-dataset values to substantiate 'near-correct' rather than leaving the interpretation to the reader.
Authors: We agree that the phrase 'near-correct splits' requires a quantitative definition to be interpretable. In the revised manuscript we will explicitly define 'near-correct' using the thresholds suggested (normalized RF < 0.05 or matching-split distance < 0.10) and add a table (or supplementary table) that reports the exact normalized RF and matching-split distances for each of the 14 datasets. The abstract will be updated to reference these concrete values rather than the current qualitative statement. revision: yes
-
Referee: [Methods] Methods (Lewis Mkv application and filtering pipeline): the model treats MAW columns as conditionally independent given the tree and branch lengths, yet each MAW is a deterministic function of the same input string; a single substitution can create or destroy multiple MAWs. The strand-aware union, entropy maximization, and parsimony capping steps are described but no diagnostic (e.g., pairwise character correlation matrix or simulation under a sequence evolution model) is provided to show that residual dependence is negligible. Because the per-branch supports and likelihood comparisons rest on this independence assumption, its violation directly undermines the claimed 'rigorous probabilistic framework'.
Authors: The referee correctly notes that MAWs are not strictly independent. The Lewis Mkv model is applied here as a practical approximation for binary characters, analogous to its use in other morphological or presence/absence datasets; the three filtering steps are intended to mitigate redundancy. No explicit diagnostic for residual dependence appears in the submitted manuscript. In revision we will add (i) a pairwise correlation matrix computed on the final character matrices of the real datasets and (ii) a small simulation study under a sequence evolution model to quantify the degree of dependence and its influence on support values. Results and any resulting caveats will be reported. revision: yes
Circularity Check
No significant circularity; derivation relies on external standards
full rationale
The paper encodes MAWs via standard suffix automata (linear-time traversal, no author-specific prior), applies the published Lewis Mkv model with ascertainment correction, and uses explicitly described filtering heuristics (strand-aware union, entropy maximization, parsimony capping). No equations, fitted parameters, or self-citations reduce the reported trees, likelihoods, or supports to quantities defined by the authors' own inputs or prior work. Evaluation uses external benchmark datasets and reference trees. The central claim therefore remains self-contained against independent components.
Axiom & Free-Parameter Ledger
axioms (2)
- standard math Suffix automaton traversal yields all minimal absent words in linear time
- domain assumption Lewis Mkv model with ascertainment bias correction is appropriate for binary character matrices derived from sequence features
Reference graph
Works this paper leans on
-
[1]
Evolutionary trees from DNA sequences: a maximum likelihood approach,
J. Felsenstein, “Evolutionary trees from DNA sequences: a maximum likelihood approach,”Journal of Molecular Evolution, vol. 17, pp. 368–376, 1981
1981
-
[2]
Alignment-free sequence analysis and applications
J. Ren, X. Bai, Y. Y. Lu, K. Tang, Y. Wang, G. Reinert, and F. Sun, “Alignment-free sequence analysis and applications.” Annual review of biomedical data science, vol. 1, pp. 93–114, 2018. [Online]. Available: https://www.semanticscholar.org/paper/ 421339791c9565581f93e395ea45fbe843af0c28
2018
-
[3]
Benchmarking of alignment-free sequence comparison methods,
A. Zielezinski, H. Z. Girgis, G. Bernard, C.-A. Leimeister, K. Tang, T. Dencker, A. Lau, S. R ¨ohling, J. Choi, M. Waterman, M. Comin, S.-H. Kim, S. Vinga, J. S. Almeida, C. Chan, B. T. James, F. Sun, B. Morgenstern, and W. Karłowski, “Benchmarking of alignment-free sequence comparison methods,”Genome Biology, vol. 20, p. null, 2019. [Online]. Available: ...
2019
-
[4]
Alignment-free sequence comparison: benefits, applications, and tools,
A. Zielezinski, S. Vinga, J. S. Almeida, and W. Karłowski, “Alignment-free sequence comparison: benefits, applications, and tools,”Genome Biology, vol. 18, p. null, 2017. [Online]. Available: https://www.semanticscholar.org/paper/ 5e3bc176613e72dac2c9665263873032e6a9bee0
2017
-
[5]
New developments of alignment-free sequence comparison: measures, statistics and next-generation sequencing,
K. Song, J. J. Ren, G. Reinert, M. Deng, M. Waterman, and F. Sun, “New developments of alignment-free sequence comparison: measures, statistics and next-generation sequencing,” Briefings in bioinformatics, vol. 15 3, pp. 343–53, 2014. [Online]. Available: https://www.semanticscholar.org/paper/ 62fe192323c5b631d95cbb5f8aa0ce844e2d03ed
2014
-
[6]
Alignment-free microbial phylogenomics under scenarios of sequence divergence, genome rearrangement and lateral genetic transfer,
G. Bernard, C. Chan, and M. Ragan, “Alignment-free microbial phylogenomics under scenarios of sequence divergence, genome rearrangement and lateral genetic transfer,”Scientific Reports, vol. 6, p. null, 2016. [Online]. Available: https://www.semanticscholar. org/paper/d55ca5e8ef293785fc7d9e71bfba79412bab5084
2016
-
[7]
Alignment-free inference of hierarchical and reticulate phylogenomic relationships,
G. Bernard, C. Chan, Y. ban Chan, X.-Y. Chua, Y. Cong, J. Hogan, S. Maetschke, and M. Ragan, “Alignment-free inference of hierarchical and reticulate phylogenomic relationships,” Briefings in Bioinformatics, vol. 20, pp. 426 – 435, 2017. [Online]. Available: https://www.semanticscholar.org/paper/ 89fd37ecb140005a9a37c5d8528b8584742f13b6
2017
-
[8]
Sequence comparison without alignment: The spam approaches,
B. Morgenstern, “Sequence comparison without alignment: The spam approaches,”bioRxiv, vol. null, p. null, 2019. [Online]. Available: https://www.semanticscholar.org/paper/ e86c211c797a9c22a49c5777bc8cdc4fa7c34b8b
2019
-
[9]
Fast alignment-free sequence comparison using spaced-word frequencies,
C.-A. Leimeister, M. Boden, S. Horwege, S. Lindner, and B. Morgenstern, “Fast alignment-free sequence comparison using spaced-word frequencies,”Bioinformatics, vol. 30, pp. 1991 – 1999, 2014. [Online]. Available: https://www.semanticscholar.org/ paper/6238641716d281d4e5e53fcfbbea3a8bbc87c079
-
[10]
Cafe: accelerated alignment-free sequence analysis,
Y. Y. Lu, K. Tang, J. J. Ren, J. Fuhrman, M. Waterman, and F. Sun, “Cafe: accelerated alignment-free sequence analysis,” Nucleic Acids Research, vol. 45, pp. W554 – W559, 2017. [Online]. Available: https://www.semanticscholar.org/paper/ 31165c3919dd0eaff05844392eeea35f459d802b
2017
-
[11]
k-mer similarity, networks of microbial genomes, and taxonomic rank,
G. Bernard, P . Greenfield, M. Ragan, and C. Chan, “k-mer similarity, networks of microbial genomes, and taxonomic rank,”mSystems, vol. 3, p. null, 2017. [Online]. Available: https://www.semanticscholar.org/paper/ ad71d87a0b67ec457a549cb8c521e205ec83ad23
2017
-
[12]
A survey and evaluations of histogram-based statistics in alignment-free sequence comparison,
B. B. Luczak, B. T. James, and H. Z. Girgis, “A survey and evaluations of histogram-based statistics in alignment-free sequence comparison,”Briefings in Bioinformatics, vol. 20, pp. 1222 – 1237, 2017. [Online]. Available: https://www.semanticscholar.org/ paper/135347386198fbdb05e2545ec9a2ae2ea87850fa
-
[13]
Fast and accurate phylogeny reconstruction using filtered spaced-word matches,
C.-A. Leimeister, S. Sohrabi-Jahromi, and B. Morgenstern, “Fast and accurate phylogeny reconstruction using filtered spaced-word matches,”Bioinformatics, vol. 33, pp. 971 – 979, 2017. [Online]. Available: https://www.semanticscholar.org/paper/ 02690b5dc7cc539b717ade150bc290e7cedd2f96
2017
-
[14]
Estimating evolutionary distances between genomic sequences from spaced-word matches,
B. Morgenstern, B. Zhu, S. Horwege, and C.-A. Leimeister, “Estimating evolutionary distances between genomic sequences from spaced-word matches,”Algorithms for Molecular Biology : AMB, vol. 10, p. null, 2015. [Online]. Available: https://www.semanticscholar.org/paper/ 17af09796b83ee525fabceef3df8630499b3c8d9
2015
-
[15]
rasbhari: Optimizing spaced seeds for database searching, read mapping and alignment-free sequence comparison,
L. Hahn, C.-A. Leimeister, and B. Morgenstern, “rasbhari: Optimizing spaced seeds for database searching, read mapping and alignment-free sequence comparison,”PLoS Computational Biology, vol. 12, p. null, 2015. [Online]. Available: https://www.semanticscholar.org/paper/ 631cafc87ea0648dc31a2a8c594be3750e95229d
2015
-
[16]
Phylogeny reconstruction based on the length distribution of k-mismatch common substrings,
B. Morgenstern, S. Sch ¨obel, and C.-A. Leimeister, “Phylogeny reconstruction based on the length distribution of k-mismatch common substrings,”Algorithms for Molecular Biology : AMB, vol. 12, p. null, 2017. [Online]. Available: https://www.semanticscholar. org/paper/0649c9e8ba3997efb4db5b68dd2bcf27a5bd78fc
2017
-
[17]
Estimating phylogenetic distances between genomic sequences based on the length distribution of k-mismatch common substrings,
B. Morgenstern, S. Schobel, and C.-A. Leimeister, “Estimating phylogenetic distances between genomic sequences based on the length distribution of k-mismatch common substrings,”arXiv: Populations and Evolution, p. null, 2017. [Online]. Available: https://www.semanticscholar.org/paper/ 8736556929e467a4ff9f39c55df36bb8eb5056ef
2017
-
[18]
kmacs: the k-mismatch average common substring approach to alignment-free sequence comparison,
C.-A. Leimeister and B. Morgenstern, “kmacs: the k-mismatch average common substring approach to alignment-free sequence comparison,”Bioinformatics, vol. 30, pp. 2000 – 2008, 2014. [Online]. Available: https://www.semanticscholar.org/paper/ f0bfa9bb4b6e1e1ffae11fa59d6fafc53e911a94
2000
-
[19]
Skmer: assembly-free and alignment-free sample identification using genome skims,
S. Sarmashghi, K. Bohmann, M. T. P . Gilbert, V . Bafna, and S. Mirarab, “Skmer: assembly-free and alignment-free sample identification using genome skims,”Genome Biology, vol. 20, p. null, 2019. [Online]. Available: https://www.semanticscholar.org/ paper/5e4425db3c8f0113054ce40812d0408afab1219e
2019
-
[20]
Phylonium: fast estimation of evolutionary distances from large samples of similar genomes,
F. Kl ¨otzl and B. Haubold, “Phylonium: fast estimation of evolutionary distances from large samples of similar genomes,”Bioinformatics, vol. 36, pp. 2040 – 2046, 2019. 11 [Online]. Available: https://www.semanticscholar.org/paper/ 262467776dccab82fd1e751e22805cf4fb475760
2040
-
[21]
Using minimal absent words to build phylogeny,
S. Chairungsee and M. Crochemore, “Using minimal absent words to build phylogeny,”Theoretical Computer Science, vol. 450, pp. 109–116, 2012
2012
-
[22]
Crochemore, C
M. Crochemore, C. Hancart, and T. Lecroq,Algorithms on Strings. Cambridge University Press, 2007
2007
-
[23]
Cd-maws: An alignment-free phylogeny estimation method using cosine distance on minimal absent word sets,
N. Anjum, R. L. Nabil, R. I. Rafi, M. S. Bayzid, and M. S. Rahman, “Cd-maws: An alignment-free phylogeny estimation method using cosine distance on minimal absent word sets,”IEEE/ACM Transactions on Computational Biology and Bioinformatics, vol. 20, pp. 196–205, 2021. [Online]. Available: https://www.semanticscholar. org/paper/4b6d6ccaa25c0e7e6aa916720fad...
2021
-
[24]
An efficient implementation of cosine distance on minimal absent word sets using suffix automata,
M. T. Ehsan, S. S. B. Mosaddek, and M. S. Rahman, “An efficient implementation of cosine distance on minimal absent word sets using suffix automata,” inProc. WALCOM: Algorithms and Computation. Springer, 2025. [Online]. Available: https: //github.com/TamimEhsan/cd-maws-sa
2025
-
[25]
An alignment-free method for phylogeny estimation using maximum likelihood,
T. Zahin, M. H. Abrar, M. Jewel, T. Tasnim, M. S. Bayzid, and A. Rahman, “An alignment-free method for phylogeny estimation using maximum likelihood,”BMC Bioinformatics, vol. 26, p. null, 2019. [Online]. Available: https://www.semanticscholar.org/ paper/33eafe7fe139c1ef0261ee0ca778c162a8db34f0
2019
-
[26]
RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies,
A. Stamatakis, “RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies,”Bioinformatics, vol. 30, no. 9, pp. 1312–1313, 2014. [Online]. Available: https://github. com/stamatak/standard-RAxML
2014
-
[27]
Sensitivity of a frequency-based alignment-free approach for phylogeny rreconstruction
Y. Hrytsenko, N. M. Daniels, and R. Schwartz, “Sensitivity of a frequency-based alignment-free approach for phylogeny rreconstruction.” 2021. [Online]. Available: https://www.semanticscholar.org/paper/ 30af5c034e2b3d417538793f3e6e281857a424de
2021
-
[28]
A likelihood approach to estimating phylogeny from discrete morphological character data,
P . O. Lewis, “A likelihood approach to estimating phylogeny from discrete morphological character data,”Systematic Biology, vol. 50, no. 6, pp. 913–925, 2001
2001
-
[29]
Comparison of phylogenetic trees,
D. F. Robinson and L. R. Foulds, “Comparison of phylogenetic trees,”Mathematical Biosciences, vol. 53, no. 1–2, pp. 131–147, 1981
1981
-
[30]
The smallest automaton recognizing the subwords of a text,
A. Blumer, J. Blumer, A. Ehrenfeucht, D. Haussler, and R. McConnell, “The smallest automaton recognizing the subwords of a text,”Theoretical Computer Science, vol. 40, pp. 31–55, 1985
1985
-
[31]
IQ-TREE 2: New models and efficient methods for phylogenetic inference in the genomic era,
B. Q. Minh, H. A. Schmidt, O. Chernomor, D. Schrempf, M. D. Woodhams, A. von Haeseler, and R. Lanfear, “IQ-TREE 2: New models and efficient methods for phylogenetic inference in the genomic era,”Molecular Biology and Evolution, vol. 37, no. 5, pp. 1530–1534, 2020. [Online]. Available: http://www.iqtree.org/
2020
-
[32]
ModelFinder: Fast model selection for accurate phylogenetic estimates,
S. Kalyaanamoorthy, B. Q. Minh, T. K. F. Wong, A. von Haeseler, and L. S. Jermiin, “ModelFinder: Fast model selection for accurate phylogenetic estimates,”Nature Methods, vol. 14, no. 6, pp. 587–589, 2017
2017
-
[33]
UFBoot2: Improving the ultrafast bootstrap approximation,
D. T. Hoang, O. Chernomor, A. von Haeseler, B. Q. Minh, and L. S. Vinh, “UFBoot2: Improving the ultrafast bootstrap approximation,” Molecular Biology and Evolution, vol. 35, no. 2, pp. 518–522, 2018
2018
-
[34]
Scientific Reports14(1), 23053 (2024)
Y. Li, L. He, R. L. He, and S. S.-T. Yau, “A novel fast vector method for genetic sequence comparison,”Scientific Reports, vol. 7, no. 1, p. 12226, 2017. [Online]. Available: https://doi.org/10.1038/s41598- 017-12493-2
-
[35]
DendroPy: a Python library for phylogenetic computing,
J. Sukumaran and M. T. Holder, “DendroPy: a Python library for phylogenetic computing,”Bioinformatics, vol. 26, no. 12, pp. 1569–1571, 2010. [Online]. Available: https://dendropy.org Papri Sahais a graduate student currently pursuing a Master of Science in Computer Science (Intelligent Systems) at the American International University-Bangladesh (AIUB), D...
2010
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.