0
Time discard rule orders population behavior for free
Conditioning as a route to stereotyped behavior in growing populations
Discarding late replication attempts favors ordered action sequences in the fastest-growing variants using only a clock and a threshold.
 full image
full image
abstract click to expand
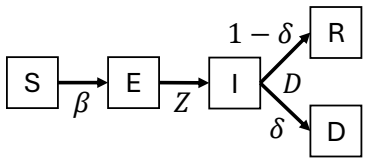
Biological systems perform complex multi-step processes in a reproducible way despite underlying stochasticity. The standard explanation is micromanagement by molecular machinery that recognizes and corrects specific errors. Here we study conditioning, a qualitatively different strategy in which attempts failing a coarse criterion are destroyed and do not leave a physical record. The surviving, i.e., conditioned, ensemble is narrower and therefore more ordered. We model conditioning through stochastic resets in a ''socks-before-shoes'' model of a growing population, where $n$ actions must be completed in any order to replicate and any replication attempt not finished by a threshold time is discarded. We find that resets impose hierarchical temporal ordering of the $n$ actions without microscopic control over which action happens when. When disorder carries a sufficient time penalty, this ordering is free: the fastest-growing population is automatically the most ordered, with no direct selection for order required. Save points, at which verified progress is preserved across resets, allow conditioning to scale to complex multi-step processes. Conditioning provides a minimal route to reliable behavior, requiring only a clock rather than molecular machinery that recognizes specific errors. For the right class of processes, it pays for itself.