An Update to Isomers of Rydberg Excitations in Argon Clusters
Pith reviewed 2026-05-23 20:54 UTC · model grok-4.3
The pith
Including a previously ignored avoided crossing in DIM calculations localizes Rydberg excitations in argon clusters on dimers rather than trimers.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
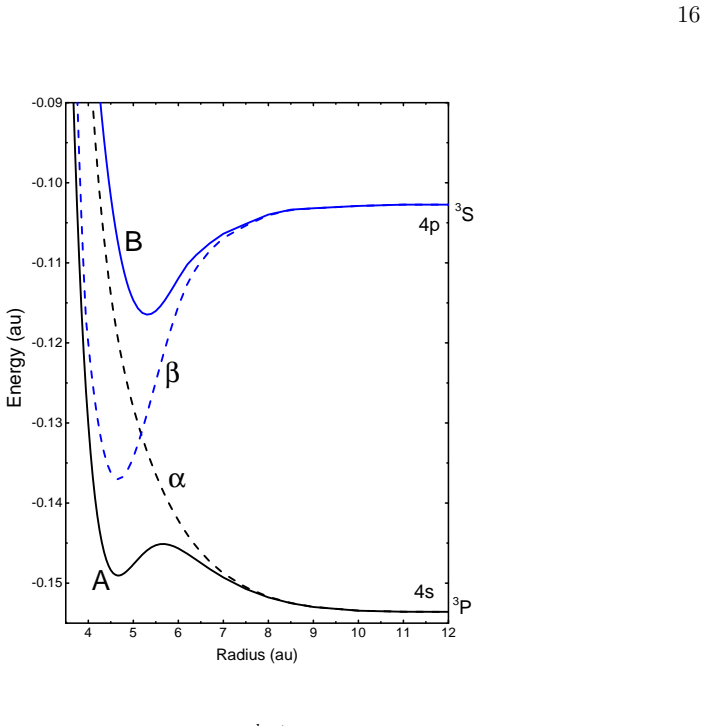
By incorporating the previously ignored strongly avoided crossing between the 3p4s and 3p4p ^{1,3}Σ states into the Diatomic-In-Molecules (DIM) method, the calculations now show the Rydberg excitation localized over a dimer (Ar₂*–Ar_{N-2}) instead of a trimer (Ar₃*–Ar_{N-3}).
What carries the argument
Diabatisation of the avoided crossing between the 3p4s and 3p4p ^{1,3}Σ states inside the DIM potential-energy surfaces for excited argon clusters.
If this is right
- The DIM and HPP methods now agree that the excitation is carried by an Ar₂* core for the lowest isomers.
- The structural motif for larger Ar_N* clusters becomes Ar₂* surrounded by neutral atoms rather than Ar₃* surrounded by neutral atoms.
- Any property derived from the isomer geometry, such as binding energies or relaxation pathways, must be recomputed with the dimer-core motif.
- The diabatisation procedure used for this crossing can be applied to other pair states in the same system.
Where Pith is reading between the lines
- Similar avoided crossings may control localization in other rare-gas cluster excitations and should be checked before trusting DIM results.
- The agreement between the two methods for argon does not yet guarantee the same localization pattern holds for neon or krypton clusters without repeating the crossing analysis.
- Experimental probes that distinguish dimer versus trimer cores, such as photoelectron spectra or fluorescence lifetimes, could now be compared directly to both methods.
Load-bearing premise
The omitted avoided crossing between the 3p4s and 3p4p ^{1,3}Σ states was the main source of the earlier difference between DIM and HPP isomer predictions.
What would settle it
A direct computation of the lowest-energy isomer geometries for a small cluster such as Ar₄* or Ar₅* using both the updated DIM surfaces and an independent high-level ab initio method, checking whether the excitation sits on two atoms or three.
Figures






read the original abstract
The effect of Diabatisation is reported in the excited argon isomers using the Diatomic-In-Molecules (DIM) method. In previous work using DIM, the lowest energy isomers of Ar$_N^*$ were shown as Ar$_3^*-$Ar$_{N-3}$, however, using the Hole-Particle-Psedopotential (HPP) method, it was shown that the excitation is localised over dimer not trimer; Ar$_2^*-$Ar$_{N-2}$. In this work we improve the DIM calculations by including previously ignored strongly avoided crossing between 3p4s and 3p4p $^{1,3}\Sigma$ states.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript updates prior DIM calculations of Rydberg-excited argon cluster isomers by incorporating the previously omitted strongly avoided crossing between the 3p4s and 3p4p ^{1,3}Σ diatomic states. This single addition is reported to reverse the localization from Ar₃*–Ar_{N-3} (as found in earlier DIM work) to Ar₂*–Ar_{N-2} (matching prior HPP results).
Significance. If the attribution holds, the result would identify a concrete, physically motivated correction that reconciles two distinct electronic-structure approaches for rare-gas Rydberg clusters and would strengthen the use of DIM for isomer energetics in this class of systems.
major comments (2)
- [Abstract] Abstract: the central claim that inclusion of the 3p4s–3p4p avoided crossing alone flips the isomer localization rests on an untested assumption that all other inputs (diatomic curves, basis, interaction terms) were identical to the earlier DIM study; no before/after comparison with only the crossing term toggled is supplied.
- [Abstract] Abstract: no numerical results, energy tables, or wave-function diagnostics are given to quantify how the avoided crossing alters the lowest isomer energies or the degree of localization; without these data the soundness of the reported shift cannot be assessed.
Simulated Author's Rebuttal
We thank the referee for their detailed review and valuable feedback on our manuscript. We address the major comments point by point below, and plan to revise the manuscript to incorporate the suggested improvements.
read point-by-point responses
-
Referee: [Abstract] Abstract: the central claim that inclusion of the 3p4s–3p4p avoided crossing alone flips the isomer localization rests on an untested assumption that all other inputs (diatomic curves, basis, interaction terms) were identical to the earlier DIM study; no before/after comparison with only the crossing term toggled is supplied.
Authors: The inputs to the DIM calculations in this work, including the diatomic curves, basis, and interaction terms, are the same as in the earlier DIM study referenced in the manuscript. To directly address the concern, we will include in the revised manuscript an explicit comparison of the isomer energies and localizations calculated with and without the avoided crossing term, while keeping all other parameters fixed. This will demonstrate the isolated effect of including the crossing. revision: yes
-
Referee: [Abstract] Abstract: no numerical results, energy tables, or wave-function diagnostics are given to quantify how the avoided crossing alters the lowest isomer energies or the degree of localization; without these data the soundness of the reported shift cannot be assessed.
Authors: We agree that the current version of the manuscript, particularly the abstract, does not include sufficient quantitative data. The full text discusses the change in localization, but to make the evidence clearer, we will add energy tables showing the lowest isomer energies for representative cluster sizes with and without the avoided crossing, along with wave-function analysis or diagnostics indicating the localization on Ar2* versus Ar3*. These additions will allow for a better assessment of the reported shift. revision: yes
Circularity Check
No circularity; update incorporates omitted physical interaction without reduction to prior outputs
full rationale
The paper reports an improvement to DIM calculations by adding the previously ignored avoided crossing between 3p4s and 3p4p states, which alters the predicted localization from trimer to dimer. No equations, fitted parameters, or self-referential derivations appear in the provided text. The change is described as inclusion of a standard physical effect rather than any re-derivation or renaming of prior results. No load-bearing self-citations, uniqueness theorems, or ansatzes are invoked. The derivation chain is therefore self-contained against external benchmarks.
Axiom & Free-Parameter Ledger
Reference graph
Works this paper leans on
-
[1]
and others [7–11]. The DIM Hamiltonian is given by; ˆHDIM = MX A MX B>A ˆHAB − (M − 2) MX A ˆHA (1) where ˆHA are the atomic fragments, ˆHAB are the diatomic fragments and M is the total number of atoms in the polyatomic molecule. The wave function for the lth electronic state solving the electronic Hamiltonian is given as a linear combination of n-electr...
-
[2]
The reasoning is similar to symmetry breaking observed in N = 4, i.e
This isomer also breaks the rotational symmetry along the excimer: the excimer is tilted in one direction as shown in Figure 5. The reasoning is similar to symmetry breaking observed in N = 4, i.e. lack of polarization in the DIM method. The Di-DIM results are summarised in table I for cluster sizes up to N=15, supplemented by the cluster size N=55 corres...
-
[3]
using DIM. In that work they neglected the strong avoided crossing which was thought to not play significant role in lowest energy isomers where they reported the excitation to be shared by a timer on the surface. In our previous work using HPP method, we showed that in the lowest energy isomers, the excitation is shared by an excimer. To determine the di...
-
[4]
F. Y. Naumkin and D. J. Wales. Rydberg excitations in rare gas clusters: Structure and electronic spectra of Ar ∗ n (3 ≤ n ≤ 25). Molecular Physics, 96(9):1295–1304, 1999
work page 1999
-
[5]
Geometry, absorption and luminescence of small Ar clusters
Mukul Dhiman, Julie Douady, and Benoit Gervais. Geometry, absorption and luminescence of small Ar clusters. Molecular Physics, page e2049905, 2022
work page 2022
-
[6]
A pseudopotential hole-particle treatment of neutral rare gas excimer systems
P Dupl` aa and F Spiegelmann. A pseudopotential hole-particle treatment of neutral rare gas excimer systems. I. Formalism. The Journal of chemical physics , 105(4):1492–1499, 1996
work page 1996
-
[7]
G Durand, P Dupl` aa, and F Spiegelmann. Pseudopotential approach of the electronic structure in clusters: application to Alkali Halides and Rare Gases. Zeitschrift f¨ ur Physik D Atoms, Molecules and Clusters , 40(1):177–181, 1997
work page 1997
-
[8]
P. Dupl` aa and F. Spiegelmann. A pseudopotential hole-particle treatment of neutral rare gas excimer systems. II. the Rydberg states of the Ar ∗ 2 dimer. Journal of Chemical Physics , 105(4):1500–1515, 1996
work page 1996
-
[9]
Frank O. Ellison. A Method of Diatomics in Molecules. I. General Theory and Application to H2O, journal = Journal of the American Chemical Society. 85(22):3540–3544, 1963
work page 1963
-
[10]
John C. Tully. Diatomics-in-molecules potential energy surfaces. II. Nonadiabatic and spin- orbit interactions. The Journal of Chemical Physics , 59(9):5135–5144, 1973
work page 1973
-
[11]
John C. Tully and Carlton M. Truesdale. Diatomics-in-molecules potential energy surfaces. III. Non-Hermitian formulation. The Journal of Chemical Physics , 65(3):1002–1007, 1976
work page 1976
-
[12]
John C. Tully. Diatomics-in-Molecules, pages 173–200. Springer US, Boston, MA, 1977
work page 1977
-
[13]
Interaction potentials ii: semiempirical atom-molecule potentials for collision the- ory
PJ Kuntz. Interaction potentials ii: semiempirical atom-molecule potentials for collision the- ory. In Atom-Molecule Collision Theory , pages 79–110. Springer, 1979
work page 1979
-
[14]
P. J. Kuntz. Interaction Potentials II: Semiempirical Atom-Molecule Potentials for Collision Theory, pages 79–110. Springer US, Boston, MA, 1979
work page 1979
-
[15]
F Spiegelmann and FX Gadea. Calcul th´ eorique en couplage Λ-Σ du spectre ´ electronique des excim` eres Ar2 et Kr2 corr´ el´ e aux configurations atomiquesnp5(n+1)s et np5(n+1)p. Journal de Physique, 45(6):1003–1023, 1984
work page 1984
-
[16]
Mukul Dhiman. Modelling excited argon clusters: ggeometries, spectroscopic properties and non-adiabatic relaxation dynamics. PhD thesis, Normandie Universit´ e, 2022
work page 2022
-
[17]
Sherin Alfalah, Daniel Kinzel, Jes´ us Gonz´ alez-V´ azquez, and Leticia Gonz´ alez. Non-adiabatic 11 photoisomerization versus photodissociation dynamics of the chiral fluoroethylene derivative (4-methylcyclohexylidene) fluoromethane. Chemical Physics, 369(2-3):138–144, 2010
work page 2010
-
[18]
Ground and excited states of Ne 2 and Ne+ 2
James S Cohen and Barry Schneider. Ground and excited states of Ne 2 and Ne+ 2 . I. Potential curves with and without spin-orbit coupling. The Journal of Chemical Physics , 61(8):3230– 3239, 1974
work page 1974
-
[19]
Semiclassical study of Ar∗ 2(3
Luc Goubert, Eric Desoppere, Willem Wieme, Rudolf Polak, Ivana Paidarova, and Gert Due Billing. Semiclassical study of Ar∗ 2(3. SIGMA. u+) excimers in a pure Ar afterglow by means of a diatomics-in-molecules potential energy surface for the Ar ∗ 3 system. The Journal of Physical Chemistry, 99(42):15479–15487, 1995
work page 1995
-
[20]
FY Naumkin and DJ Wales. Diatomics-in-molecules potentials incorporating ab initio data: Application to ionic, Rydberg-excited, and molecule-doped rare gas clusters.Computer physics communications, 145(1):141–155, 2002. 12 V. APPENDIX: SUPPLEMENTARY MATERIAL A. Application to 3p54s excited configuration To get 3p54s excited state, we put x† = 4s, so we ca...
work page 2002
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.