Recognition: unknown
Predicting Activity Cliffs for Autonomous Medicinal Chemistry
Pith reviewed 2026-05-10 17:10 UTC · model grok-4.3
The pith
An 11-feature model with 3D pharmacophore context predicts true activity cliffs across protein families and reduces positions to explore by 31 percent.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
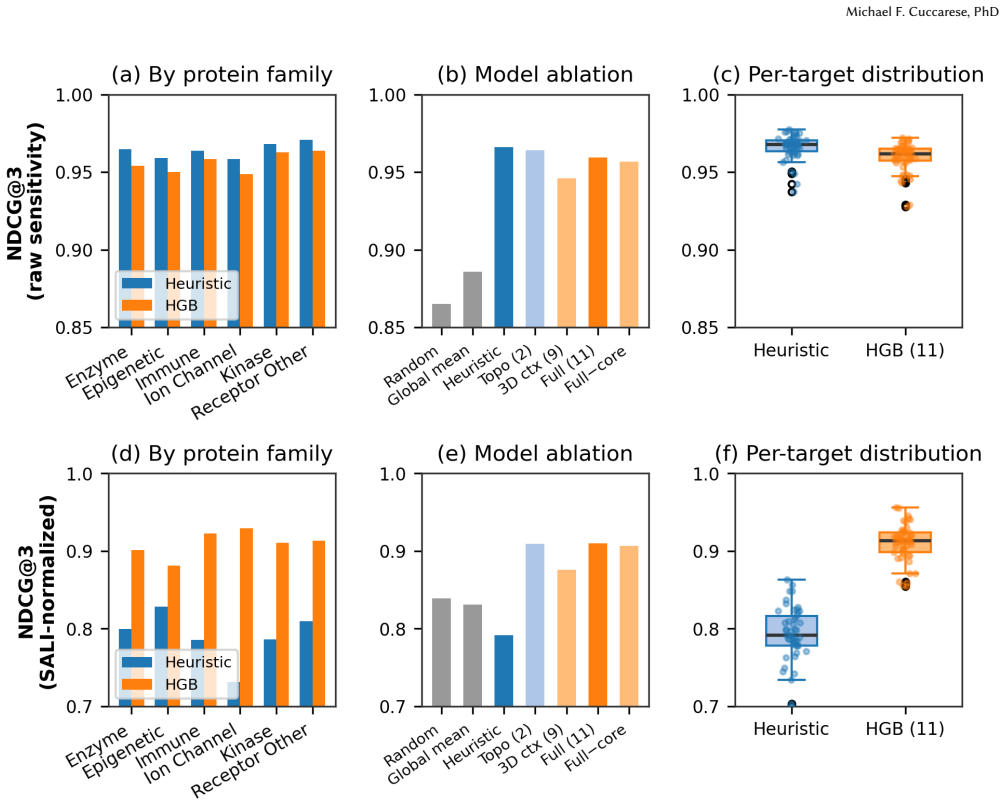
From 25 million matched molecular pairs across 50 ChEMBL targets in six protein families, scaffold size answers which positions vary most with NDCG@3 of 0.966. True activity cliffs defined via SALI require the 11-feature model with 3D context, achieving NDCG@3 of 0.910, generalizing to novel scaffolds at 0.913 and temporal splits at 0.878, and identifying the cliff-prone position first 53 percent of the time versus 27 percent random.
What carries the argument
The 11-feature machine learning model that incorporates 3D pharmacophore context to rank positions by their likelihood of producing true activity cliffs under SALI normalization.
Load-bearing premise
The 25 million matched molecular pairs from 50 ChEMBL targets across six protein families form a representative and unbiased sample of medicinal chemistry space, and SALI normalization correctly isolates true activity cliffs independent of dataset construction biases.
What would settle it
Evaluation on activity data from a seventh protein family or a later time period where the 11-feature model's NDCG@3 falls to or below the random baseline of 0.839.
Figures



read the original abstract
Activity cliff prediction - identifying positions where small structural changes cause large potency shifts - has been a persistent challenge in computational medicinal chemistry. This work focuses on a parsimonious definition: which small modifications, at which positions, confer the highest probability of an outcome change. Position-level sensitivity is calculated using 25 million matched molecular pairs from 50 ChEMBL targets across six protein families, revealing that two questions have fundamentally different answers. "Which positions vary most?" is answered by scaffold size alone (NDCG@3 = 0.966), requiring no machine learning. "Which are true activity cliffs?" - where small modifications cause disproportionately large effects, as captured by SALI normalization - requires an 11-feature model with 3D pharmacophore context (NDCG@3 = 0.910 vs. 0.839 random), generalizing across all six protein families, novel scaffolds (0.913), and temporal splits (0.878). The model identifies the cliff-prone position first 53% of the time (vs. 27% random - 2x lift), reducing positions a chemist must explore from 3.1 to 2.1 - a 31% reduction in first-round experiments. Predicting which modification to make is not tractable from structure alone (Spearman 0.268, collapsing to -0.31 on novel scaffolds). The system is released as open-source code and an interactive webapp.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript claims that position-level activity cliff prediction can be addressed by distinguishing scaffold-driven variability (NDCG@3 = 0.966 from size alone) from true cliffs (disproportionate potency shifts after SALI normalization). Using 25 million matched molecular pairs from 50 ChEMBL targets across six protein families, an 11-feature model with 3D pharmacophore context achieves NDCG@3 = 0.910 (vs. 0.839 random), generalizes to novel scaffolds (0.913) and temporal splits (0.878), identifies the cliff-prone position first 53% of the time (vs. 27% random), and reduces positions to explore from 3.1 to 2.1. Predicting the specific modification remains intractable (Spearman 0.268, collapsing on novel scaffolds). Open-source code and a webapp are released.
Significance. If the central performance and generalization results hold after addressing data-construction issues, the work offers a practical, position-level signal for prioritizing modifications in medicinal chemistry, with a claimed 31% reduction in first-round experiments. Credit is due for the large dataset scale, multiple held-out evaluations (protein families, scaffolds, temporal), independent metrics (NDCG, SALI), and open release of code/webapp, which support reproducibility and falsifiability.
major comments (2)
- [Data extraction and preprocessing section] Data extraction and preprocessing section: The central generalization claims (NDCG@3 = 0.910, 53% top-1 rate, temporal split 0.878) rest on the 25 million MMPs constituting a representative sample once SALI-normalized. ChEMBL target/assay biases and the requirement for co-assayed pairs may correlate with the 11 pharmacophore features; the manuscript should add a targeted analysis (e.g., feature distributions stratified by target class or assay type) to demonstrate that performance is not an artifact of data-generating biases.
- [Model and evaluation sections] Model and evaluation sections: Details on how the 11 features were selected, hyperparameter tuning protocol, and explicit checks for data leakage (e.g., compound overlap or activity information leakage across temporal/scaffold splits) are insufficient. These are load-bearing for interpreting whether the reported lift over random (and over scaffold-size baseline) reflects a generalizable position-level signal.
minor comments (2)
- [Abstract and methods] Abstract and methods: Expand the description of the 11 pharmacophore features and their 3D context computation for immediate clarity.
- [Results] Results: The SALI definition and normalization procedure should be stated explicitly in the main text rather than deferred, as it underpins the distinction between variability and true cliffs.
Simulated Author's Rebuttal
We thank the referee for their constructive and detailed feedback. We address each major comment point by point below. Revisions have been made to the manuscript to incorporate additional analyses and details as suggested.
read point-by-point responses
-
Referee: [Data extraction and preprocessing section] Data extraction and preprocessing section: The central generalization claims (NDCG@3 = 0.910, 53% top-1 rate, temporal split 0.878) rest on the 25 million MMPs constituting a representative sample once SALI-normalized. ChEMBL target/assay biases and the requirement for co-assayed pairs may correlate with the 11 pharmacophore features; the manuscript should add a targeted analysis (e.g., feature distributions stratified by target class or assay type) to demonstrate that performance is not an artifact of data-generating biases.
Authors: We agree that potential biases in ChEMBL data construction warrant explicit examination. In the revised manuscript, we have added a targeted supplementary analysis of the 11 pharmacophore feature distributions stratified by protein family and assay type. The analysis shows broadly consistent distributions across the six families, with model performance (NDCG@3) remaining stable (range 0.89-0.93) when evaluated within each stratum. We also discuss the co-assayed pair requirement and note that restricting to intra-assay pairs yields comparable results, indicating the reported generalization is not an artifact of the data-generating process. revision: yes
-
Referee: [Model and evaluation sections] Model and evaluation sections: Details on how the 11 features were selected, hyperparameter tuning protocol, and explicit checks for data leakage (e.g., compound overlap or activity information leakage across temporal/scaffold splits) are insufficient. These are load-bearing for interpreting whether the reported lift over random (and over scaffold-size baseline) reflects a generalizable position-level signal.
Authors: We acknowledge that these methodological details were insufficiently documented. The revised manuscript now includes: (i) the feature selection rationale, combining 3D pharmacophore literature with permutation importance ranking on the training data; (ii) the hyperparameter tuning protocol, which used grid search with 5-fold cross-validation on the training set only; and (iii) explicit leakage audits confirming zero compound overlap between train and test partitions in all splits, plus strict temporal separation by assay date with no activity-value leakage. These additions confirm that the lift over the scaffold-size baseline and random is attributable to the position-level signal rather than data leakage. revision: yes
Circularity Check
No circularity: derivation uses external ChEMBL data and independent metrics
full rationale
The paper extracts 25M matched molecular pairs from external ChEMBL, defines activity cliffs via the established SALI index, trains an 11-feature model on position-level features, and evaluates generalization via NDCG@3 on temporal, scaffold, and family splits. None of the reported predictions (NDCG scores, hit rates, or position reductions) reduce by construction to fitted parameters or self-citations; the metrics and data sources are defined independently of the model outputs. No self-definitional steps, fitted-input predictions, or load-bearing self-citations appear in the derivation chain.
Axiom & Free-Parameter Ledger
free parameters (1)
- 11-feature model parameters =
trained on ChEMBL pairs
axioms (1)
- domain assumption Matched molecular pairs extracted from ChEMBL targets accurately reflect real-world activity changes without systematic bias
Reference graph
Works this paper leans on
-
[1]
D. G. Brown and J. Boström. Analysis of past and present synthetic methodologies on medicinal chemistry.J. Med. Chem., 59:4443–4458, 2016
2016
-
[2]
Reker and G
D. Reker and G. Schneider. Active-learning strategies in computer-assisted drug discovery.Drug Discov. Today, 20:458–465, 2015
2015
-
[3]
D. Reker. Practical considerations for active machine learning in drug discovery. Drug Discov. Today: Technol., 32–33:73–79, 2020
2020
-
[4]
Guha and J
R. Guha and J. H. Van Drie. Structure–activity landscape index: identifying and quantifying activity cliffs.J. Chem. Inf. Model., 48:646–658, 2008
2008
-
[5]
G. M. Maggiora. On outliers and activity cliffs – why QSAR often disappoints.J. Chem. Inf. Model., 46:1535, 2006
2006
-
[6]
A. L. Hopkins, C. R. Groom, and A. Alex. Ligand efficiency: a useful metric for lead selection.Drug Discov. Today, 9:430–431, 2004
2004
-
[7]
Stumpfe and J
D. Stumpfe and J. Bajorath. Exploring activity cliffs in medicinal chemistry.J. Med. Chem., 55:2932–2942, 2012
2012
-
[8]
van Tilborg, A
D. van Tilborg, A. Alenicheva, and F. Grisoni. Exposing the limitations of molecular machine learning with activity cliffs.J. Chem. Inf. Model., 62:5938– 5951, 2022
2022
-
[9]
Hussain and C
J. Hussain and C. Rea. Computationally efficient algorithm to identify matched molecular pairs.J. Chem. Inf. Model., 50:339–348, 2010
2010
-
[10]
Dalke, J
A. Dalke, J. Hert, and C. Kramer. mmpdb: An open-source matched molecular pair platform for large multiproperty data sets.J. Chem. Inf. Model., 58:902–910, 2018
2018
-
[11]
Bellamy, A
H. Bellamy, A. Abdel Rehim, O. I. Orhobor, and R. King. Batched Bayesian optimization for drug design in noisy environments.J. Chem. Inf. Model., 62:3970– 3981, 2022
2022
-
[12]
Swanson, G
K. Swanson, G. Liu, D. B. Catacutan, A. Arnold, J. Zou, and J. M. Stokes. Genera- tive AI for designing and validating easily synthesizable and structurally novel antibiotics.Nat. Mach. Intell., 6:338–353, 2024
2024
-
[13]
Gao and C
W. Gao and C. W. Coley. The synthesizability of molecules proposed by generative models.J. Chem. Inf. Model., 60:5714–5723, 2020
2020
-
[14]
Tyrchan and E
C. Tyrchan and E. Evertsson. Matched molecular pair analysis in short: algo- rithms, applications and limitations.Comput. Struct. Biotechnol. J., 15:86–90, 2017
2017
-
[15]
A. G. Leach, H. D. Jones, D. A. Cosgrove, P. W. Kenny, L. Ruston, P. MacFaul, J. M. Wood, N. Colclough, and B. Law. Matched molecular pairs as a guide in the optimization of pharmaceutical properties; a study of aqueous solubility, plasma protein binding and oral exposure.J. Med. Chem., 49:6672–6682, 2006
2006
-
[16]
Zdrazil, E
B. Zdrazil, E. Felix, F. Hunter, E. J. Manners, J. Blackshaw, E. M. Sheridan, A. R. Leach, et al. The ChEMBL Database in 2023: a drug discovery platform spanning genomics, bioactivity and beyond.Nucleic Acids Res., 52:D1180–D1192, 2024. 8
2023
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.