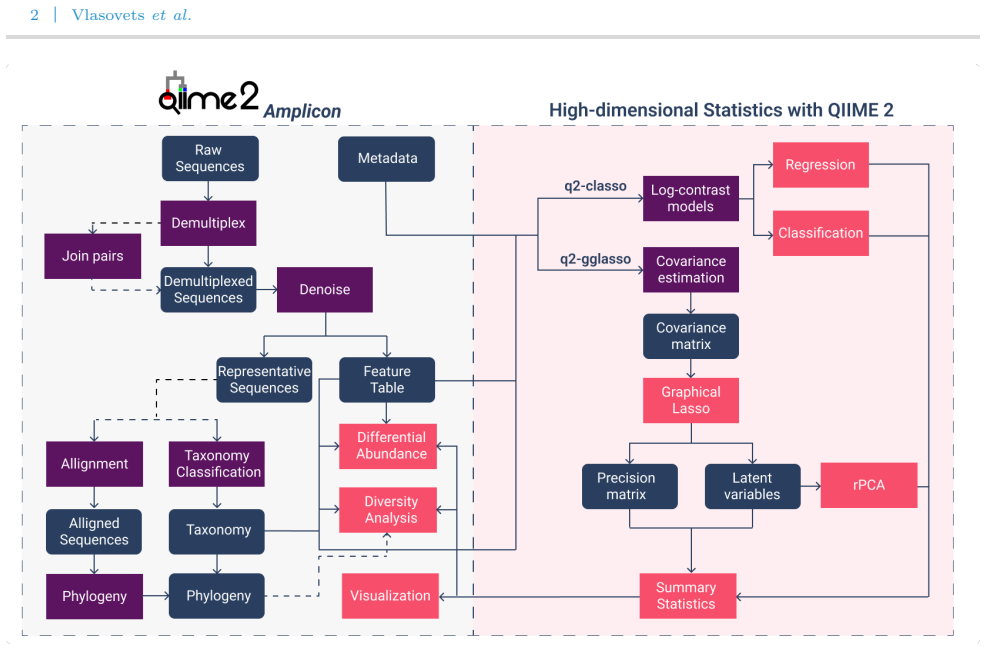
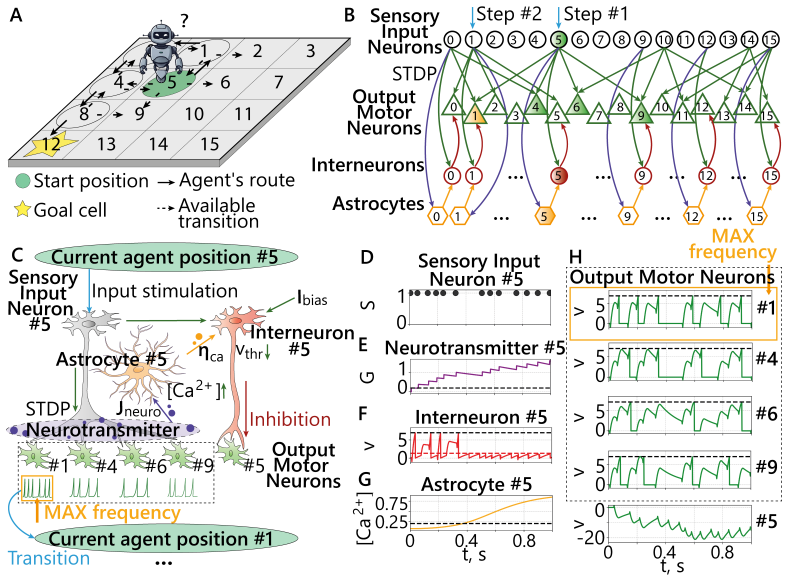
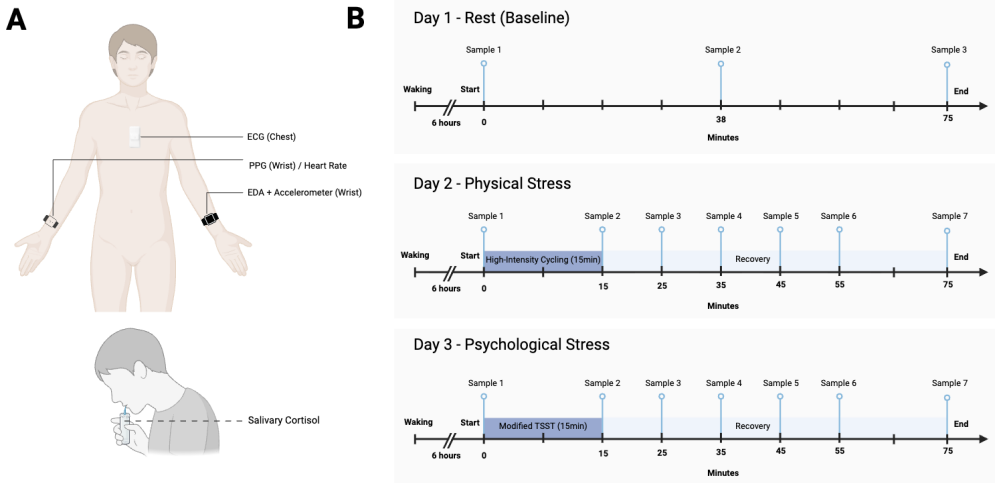
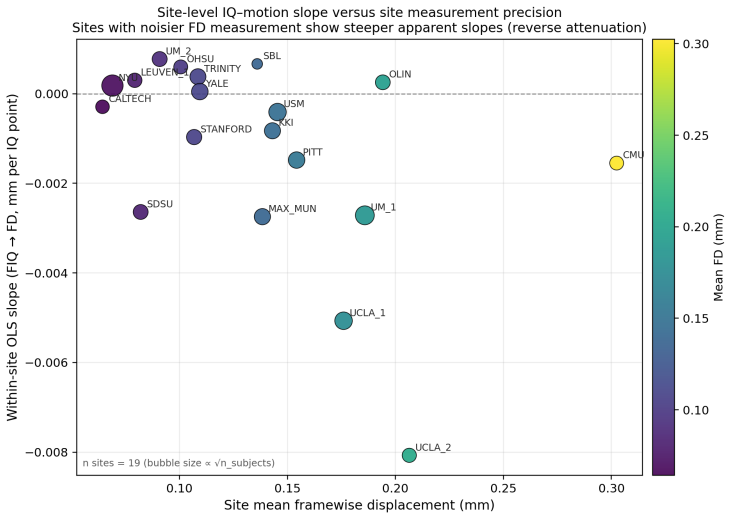
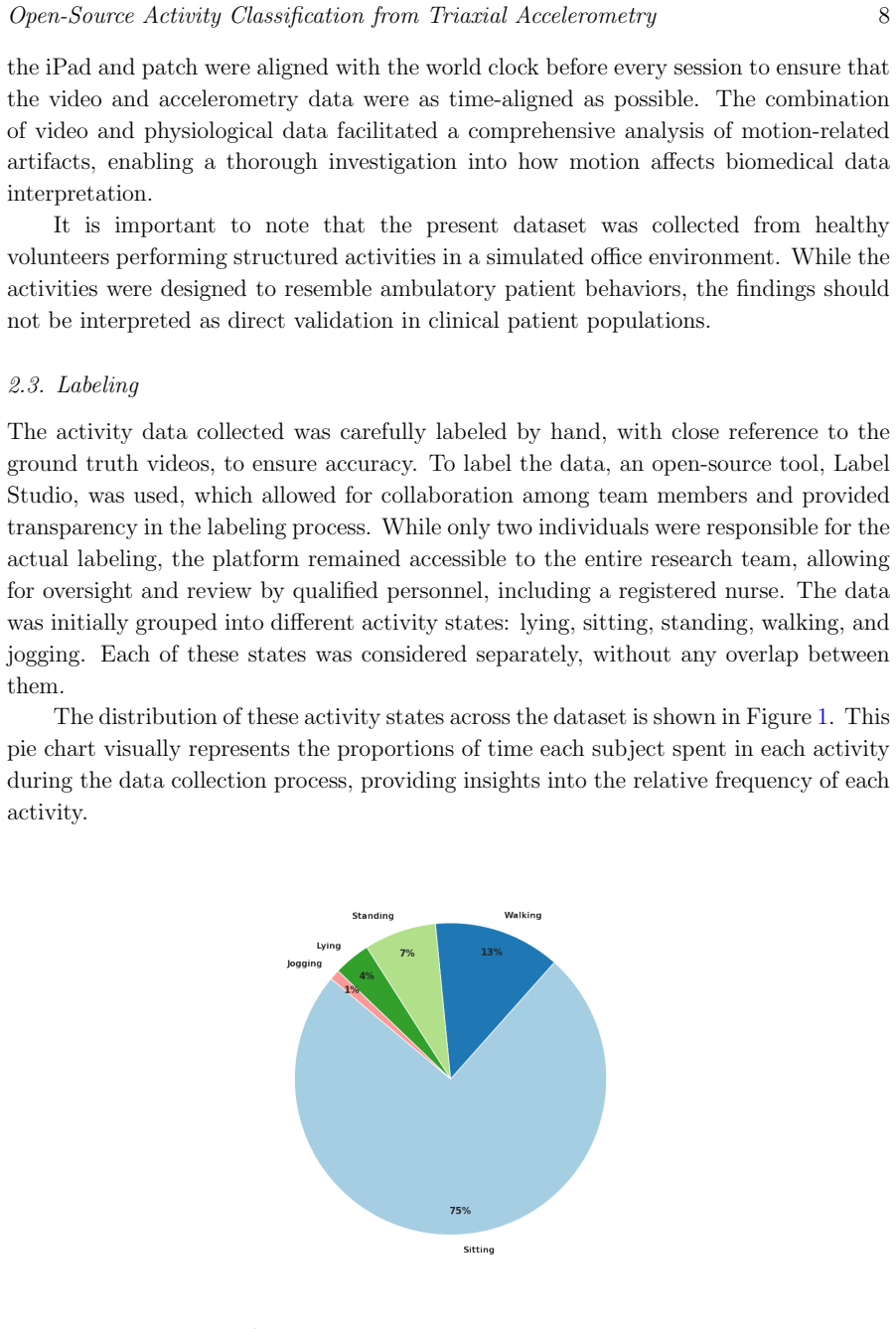
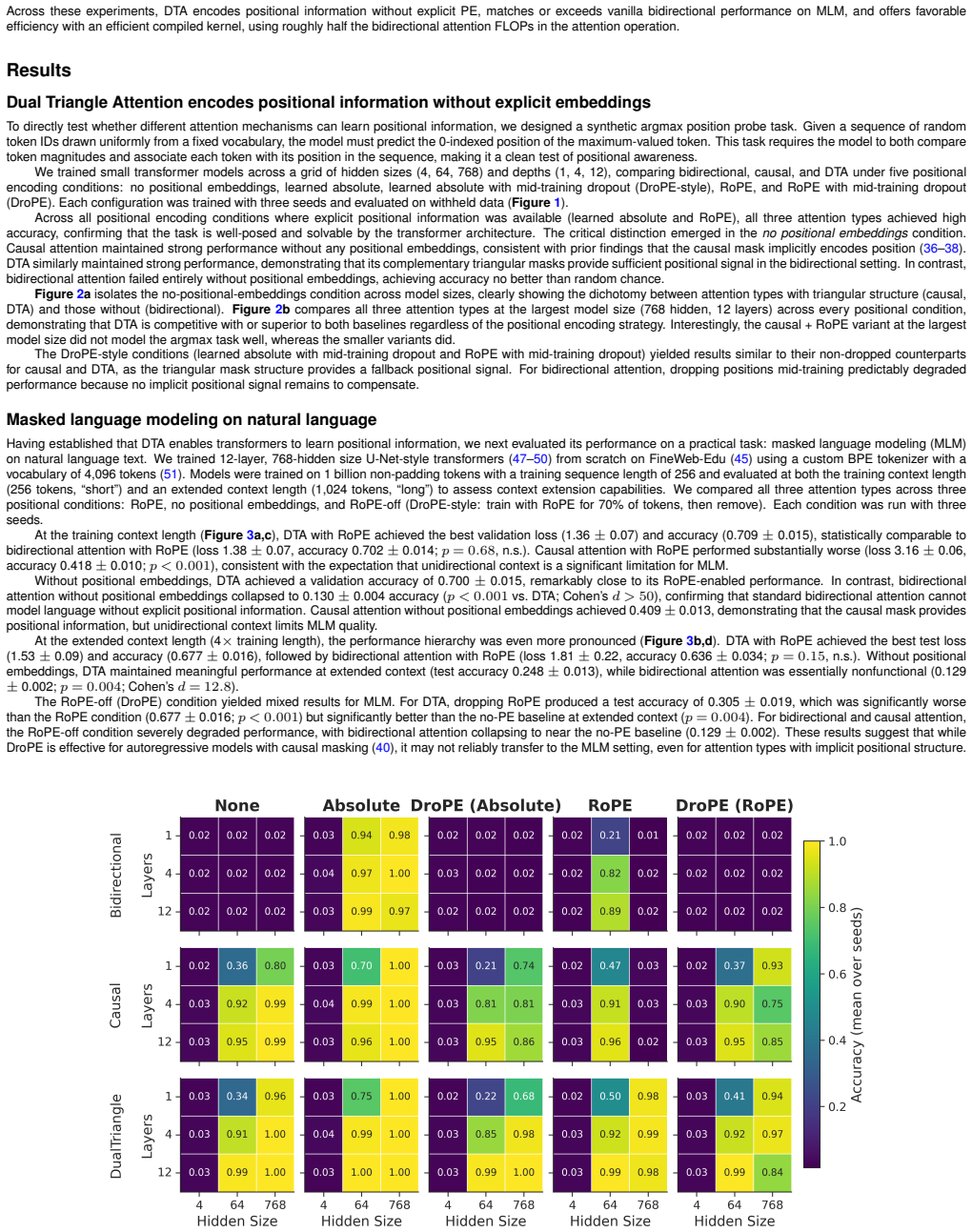
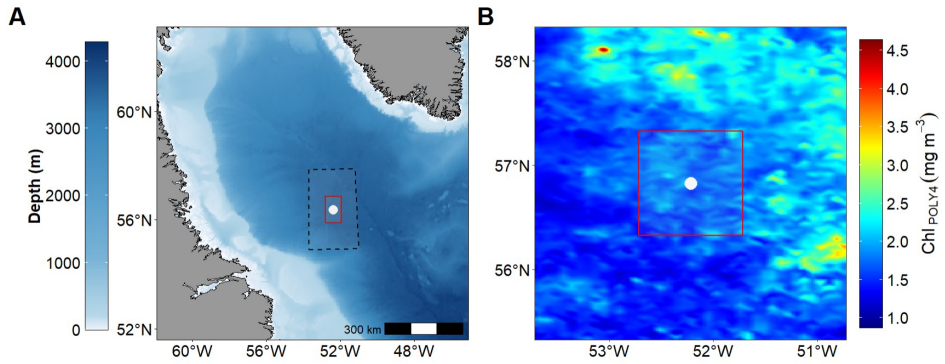
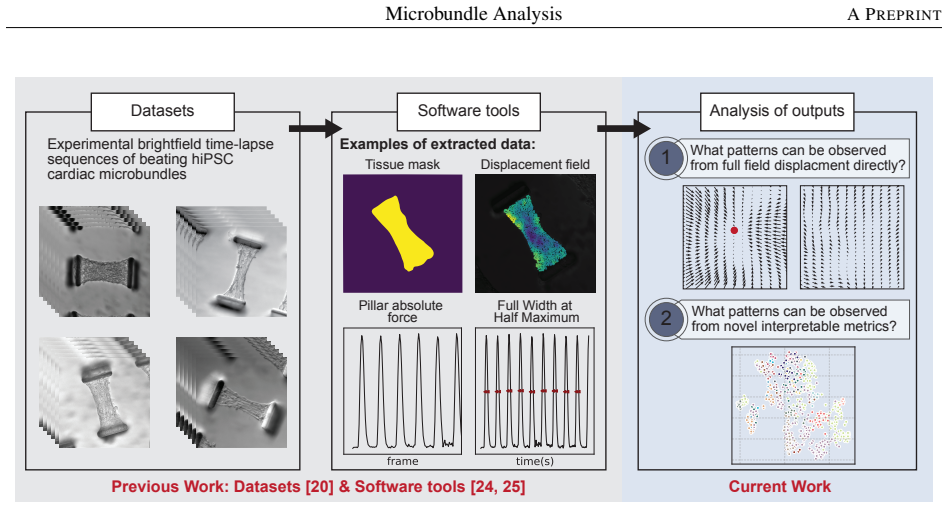
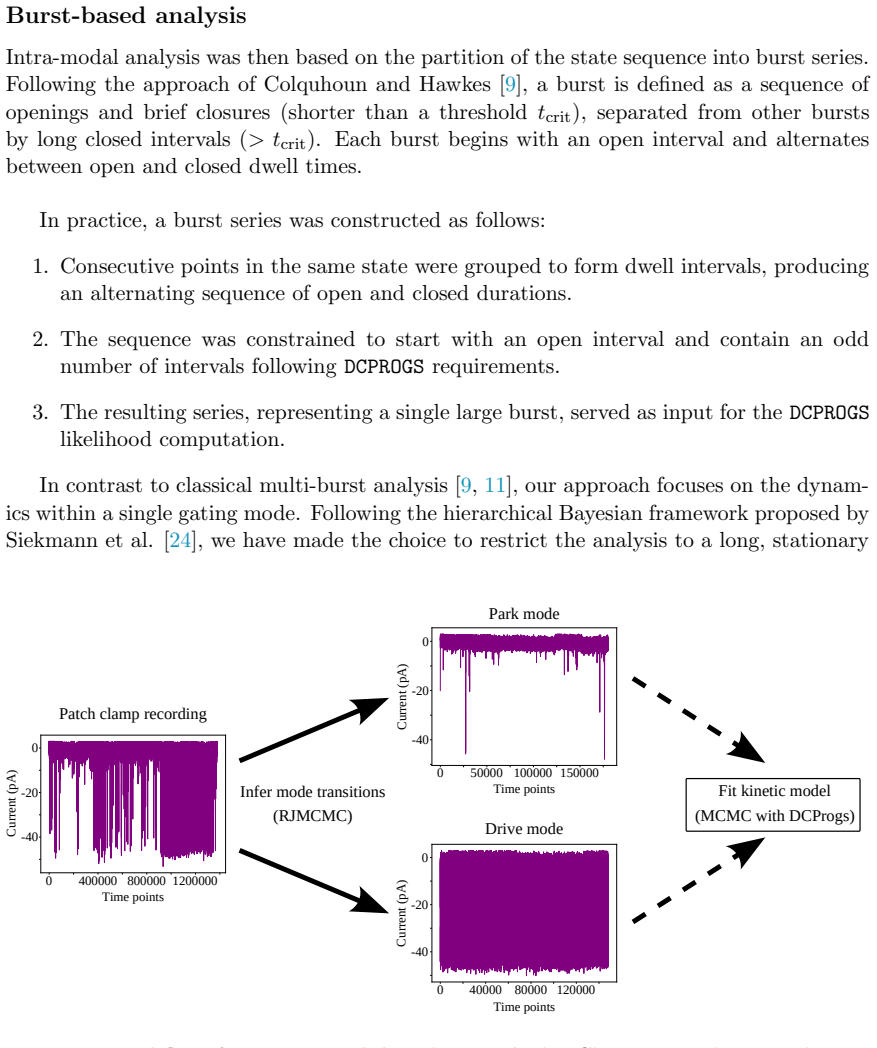
0
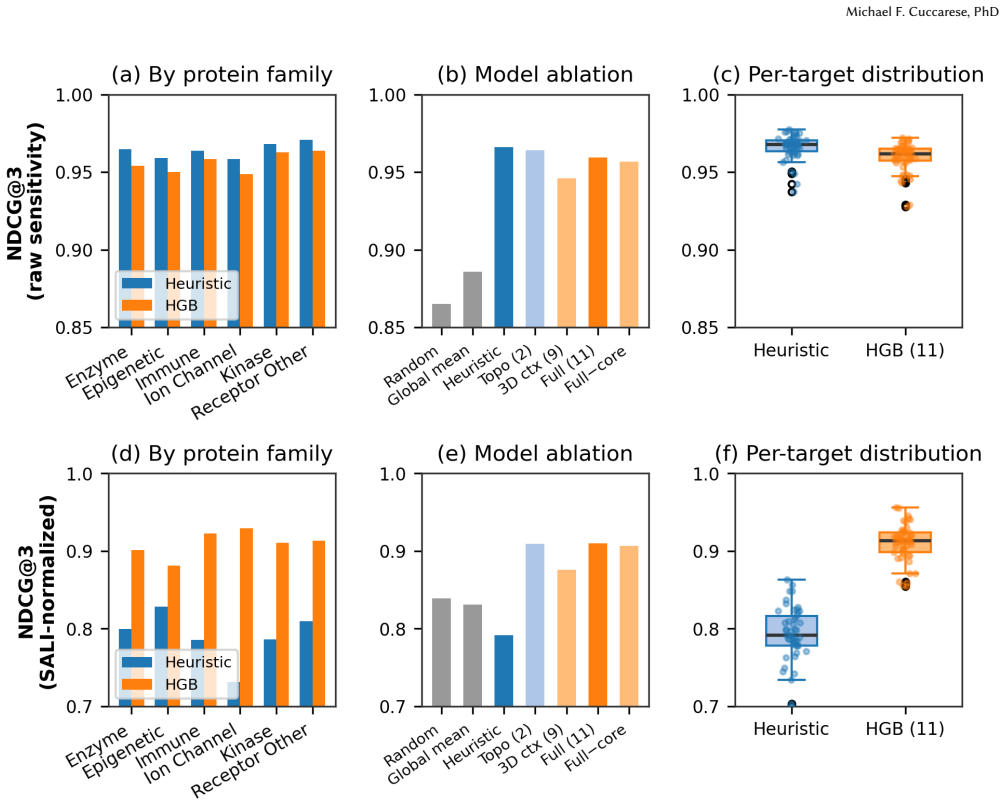
Missed events correction clarifies IP3R Park and Drive modes
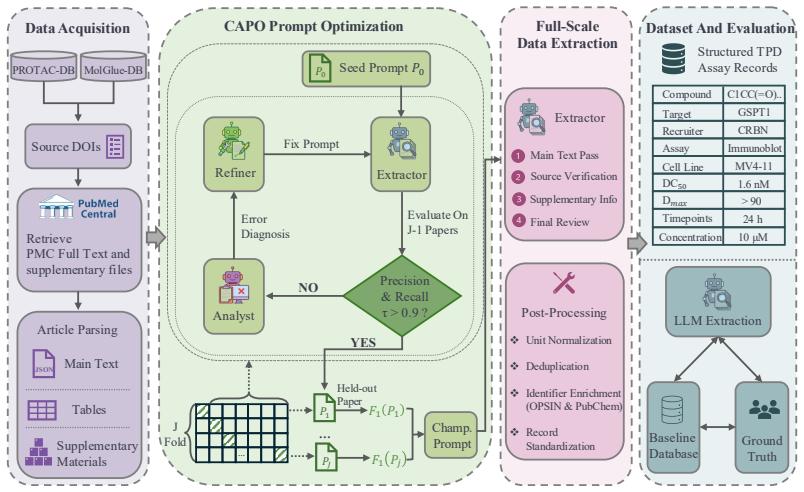
Accounting for Missed Events in the Bayesian Modeling of IP3R Multimodal Gating
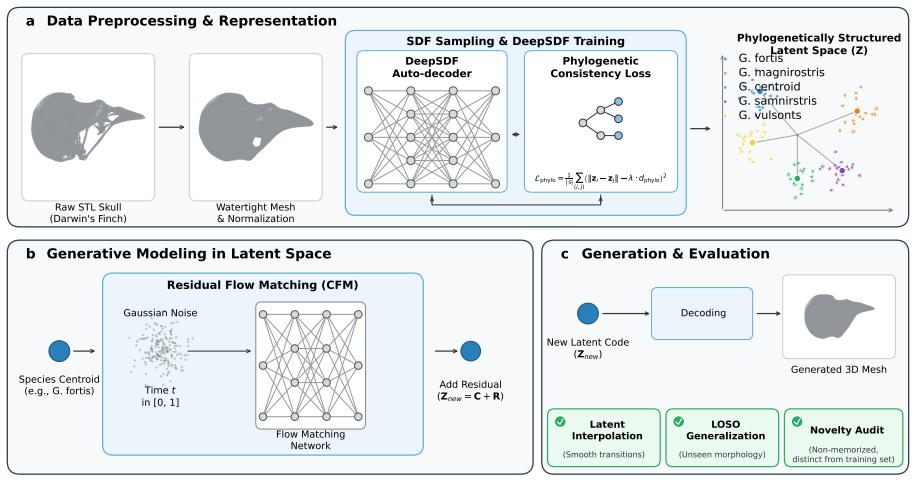
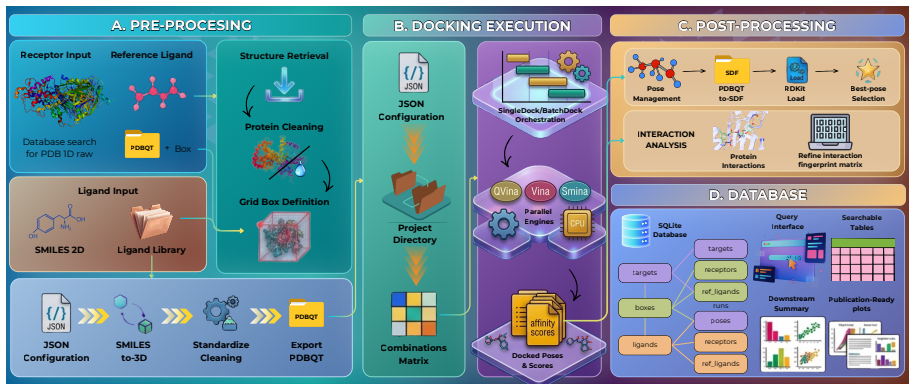
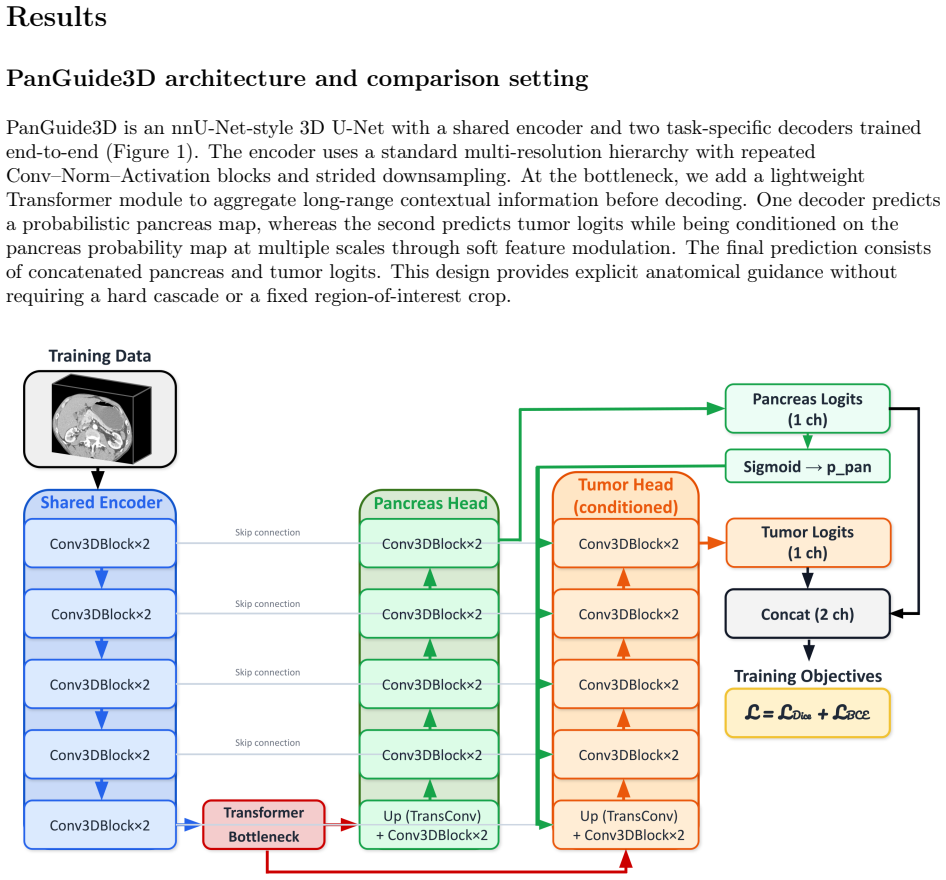
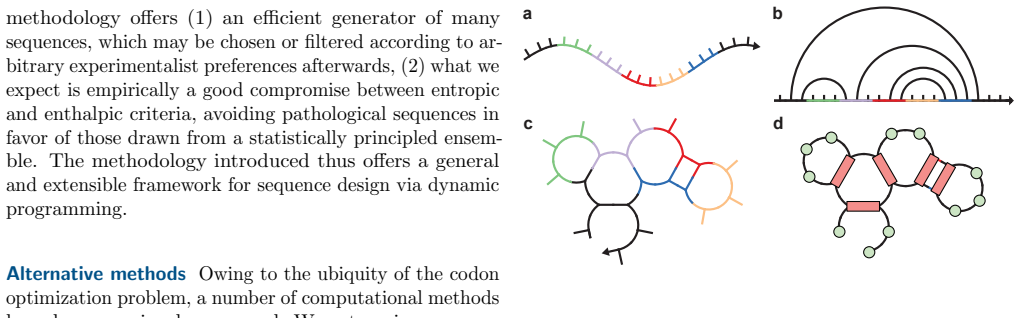
Bayesian analysis reveals both modes use the same 3-state model but stabilize different closed states, with calcium controlling switches.
 full image
full image
abstract click to expand
The Inositol 1,4,5-trisphosphate receptor channel (IP 3 R) is an important calcium channel involved in calcium-induced calcium release, playing a prominent role in intracellular calcium signaling. However, accurately characterizing its gating behavior remains a challenge, particularly due to the temporal resolution of patch clamp techniques that is not large enough to detect all short-lived events. This limitation can significantly bias the inference of kinetic models describing the receptor activity. To address this issue, we focused on the quantitative analysis of IP 3 R gating behavior using patch clamp data, with particular attention to missed events. We modeled IP 3 R channel gating using Hierarchical Markov chains and used a Bayesian approach that integrates missed event correction directly into the likelihood function, enabling more accurate parameter inference and model evaluation. We show that accounting for missed events deeply clarifies the multi-modal model that emerges from model selection. In this new model, the Park and Drive modes both consist of the same 3-state Markov model, with mode-dependent kinetic parameters: the Drive mode stabilizes the closed state directly connected to the open one, whereas the Park mode stabilizes the other closed state, that is not connected to the open one. Intermediate Ca 2+ concentrations are found to strongly depress the Drive to Park transition rate, so that the IP 3 R channel undergoes frequent transitions to the Park mode only for __ 50 nM or micromolar Ca 2+ concentrations. Overall, our approach provides a refined perspective on IP 3 R channel modeling and highlights the critical importance of accounting for missed events upon model selection based on single-channel recordings.