Recognition: unknown
Neutralization titers reveal the structure of polyclonal antibody responses
Pith reviewed 2026-05-10 15:54 UTC · model grok-4.3
The pith
The statistics of neutralization titers alone suffice to infer the composition of polyclonal antibody responses to pathogens like influenza.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
The composition of a polyclonal antibody response, which is difficult to measure directly, can be quantitatively predicted from the statistics of neutralization titers. In a cohort responding to influenza, the response may be collective, involving many antibodies, or dominated by few strong binders, leading to a broad distribution of titers across individuals that follows a Gumbel distribution from extreme value theory. An equilibrium binding model captures the titer data and illustrates the underlying structure, with pre-challenge titers also fitting Gumbel statistics. This framework extends to responses against other pathogens.
What carries the argument
The Gumbel distribution arising from extreme value theory when titers are dominated by the strongest binders, together with an equilibrium binding model that links antibody affinity and concentration to neutralization outcomes.
If this is right
- Titer variation across individuals encodes information about the number and strength distribution of antibodies in the response.
- Responses can be classified as collective or dominated by few binders based on the shape of the titer distribution.
- Pre-immune challenge titers follow the same Gumbel statistics as post-challenge in some cohorts.
- The equilibrium model allows quantitative predictions without experimental isolation of antibodies.
- The approach applies generically to other pathogens beyond influenza.
Where Pith is reading between the lines
- Such titer-based inference could allow large-scale population studies of immune response structure without costly molecular techniques.
- It might help explain why some individuals show broader protection against viral variants based on whether their response is collective.
- Testing the model on responses to different viruses could reveal if collective responses are more common for certain pathogens.
Load-bearing premise
Titer differences between individuals primarily reflect variations in the underlying antibody populations rather than being dominated by measurement errors, genetic variations, or dynamic effects outside the equilibrium model.
What would settle it
Finding that the predicted number of strong antibodies from titer distributions does not match the actual diversity and affinities measured by sequencing and expressing monoclonal antibodies from the same individuals would falsify the claim.
Figures



read the original abstract
The composition of a polyclonal antibody response is hard to measure experimentally but contains vital information about the robustness of immunity. Here, we argue that the statistics of neutralization titers alone can be used to make quantitative predictions about the composition of the response, circumventing challenges arising through sequencing and monoclonal antibody expression. We show that the response against influenza within a cohort can be either driven by a collective phenomenon where many antibodies contribute to neutralization, or dominated by just a few strong binders, leading to a broad distribution of titers across individuals described by a Gumbel distribution from extreme value theory. Comparing titers across cohorts, we find that Gumbel statistics {accurately describe} individuals prior to an immune challenge. We propose an equilibrium binding model that quantitatively captures titer data and illustrates the structure of the polyclonal response. Our approach extends generically to immune responses to other pathogens.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript argues that neutralization titer statistics alone can be used to make quantitative predictions about the composition of polyclonal antibody responses, circumventing sequencing and mAb expression challenges. It claims that influenza responses within a cohort are either collective (many antibodies contributing to neutralization) or dominated by a few strong binders, producing broad titer distributions across individuals that follow a Gumbel distribution from extreme value theory. An equilibrium binding model is proposed that quantitatively captures the titer data and illustrates response structure, with the approach asserted to extend generically to other pathogens.
Significance. If the central claims hold, this would provide a significant non-invasive method to infer polyclonal response details (collective vs. few-dominant) directly from measurable titers, with implications for understanding immunity robustness and vaccine responses. The link to extreme value theory for explaining titer variation across individuals is a theoretically appealing contribution. The equilibrium model offers a potential framework for mapping titers to antibody number and affinity distributions.
major comments (3)
- [Abstract] Abstract: The assertion that the equilibrium binding model 'quantitatively captures titer data' and enables predictions about response composition is load-bearing for the central claim, yet the text provides no details on whether model parameters (e.g., antibody affinities or numbers) are determined independently of the cohort titer observations or fitted to the same data, raising a circularity concern that must be resolved with explicit equations and fitting procedures.
- [Cohort analysis] Cohort analysis and model sections: The inference that titer variation encodes collective vs. few-dominant antibody contributions (via the proposed mapping to Gumbel statistics) lacks any cross-validation against independent compositional data such as sequenced repertoires or expressed monoclonal antibody affinities; without this, the claim that titers 'reveal the structure' cannot be substantiated and remains vulnerable to confounding from measurement noise or non-equilibrium effects.
- [Results] Gumbel distribution claim: The statement that Gumbel statistics accurately describe pre-challenge individuals is central to distinguishing response types, but no specific goodness-of-fit metrics, parameter estimates, or comparisons to alternative distributions (e.g., log-normal) are reported to support the extreme value theory application.
minor comments (2)
- [Abstract] Abstract contains a formatting artifact ('Gumbel statistics {accurately describe}') that should be corrected for clarity.
- [Model] Notation for titer values (e.g., IC50) and model variables could be defined more explicitly in the model section to aid readability.
Simulated Author's Rebuttal
We thank the referee for their constructive comments on our manuscript. These have prompted us to clarify key aspects of our analysis and model. We respond to each major comment in turn below, indicating where revisions will be made to the manuscript.
read point-by-point responses
-
Referee: [Abstract] Abstract: The assertion that the equilibrium binding model 'quantitatively captures titer data' and enables predictions about response composition is load-bearing for the central claim, yet the text provides no details on whether model parameters (e.g., antibody affinities or numbers) are determined independently of the cohort titer observations or fitted to the same data, raising a circularity concern that must be resolved with explicit equations and fitting procedures.
Authors: We agree that additional details are needed to address potential concerns about circularity. In the revised manuscript, we will expand the methods section to include the explicit equations of the equilibrium binding model. The model parameters, including the mean and variance of log-affinity distributions and the typical number of antibodies, are drawn from independent experimental literature on monoclonal antibody affinities and polyclonal response sizes (e.g., from prior studies on influenza HA binding). These are not fitted to the titer cohort data; instead, the model generates predicted titer distributions that are then compared to observations. We will also report the specific parameter values used and any sensitivity analyses performed. revision: yes
-
Referee: [Cohort analysis] Cohort analysis and model sections: The inference that titer variation encodes collective vs. few-dominant antibody contributions (via the proposed mapping to Gumbel statistics) lacks any cross-validation against independent compositional data such as sequenced repertoires or expressed monoclonal antibody affinities; without this, the claim that titers 'reveal the structure' cannot be substantiated and remains vulnerable to confounding from measurement noise or non-equilibrium effects.
Authors: The manuscript's primary aim is to demonstrate that titer statistics can provide quantitative insights into response composition without the need for direct compositional measurements, which are technically challenging and not always available. While we acknowledge that cross-validation with sequencing or mAb data would be valuable for further confirmation, such data are not part of the current study and obtaining them would require a separate experimental effort beyond the scope of this work. We will add a new paragraph in the discussion to explicitly address potential confounding factors, including measurement noise in titer assays and deviations from equilibrium assumptions, and outline how the Gumbel framework could be tested in future combined studies. The theoretical derivation from extreme value theory provides an independent rationale for the observed distributions under the collective or few-dominant scenarios. revision: partial
-
Referee: [Results] Gumbel distribution claim: The statement that Gumbel statistics accurately describe pre-challenge individuals is central to distinguishing response types, but no specific goodness-of-fit metrics, parameter estimates, or comparisons to alternative distributions (e.g., log-normal) are reported to support the extreme value theory application.
Authors: We will revise the results and supplementary materials to include quantitative assessments of the Gumbel fit. Specifically, we will report the estimated location and scale parameters for the Gumbel distributions fitted to pre-challenge titer data from each cohort. Goodness-of-fit will be evaluated using the Kolmogorov-Smirnov statistic and p-values, as well as comparisons to log-normal and other distributions via Akaike information criterion (AIC) or likelihood ratio tests. These additions will provide rigorous statistical support for the application of extreme value theory. revision: yes
Circularity Check
Equilibrium binding model fitted to titer data used to claim quantitative predictions of polyclonal structure
specific steps
-
fitted input called prediction
[Abstract]
"We propose an equilibrium binding model that quantitatively captures titer data and illustrates the structure of the polyclonal response."
The model is calibrated ('captures') to the observed titer statistics; the same calibrated model is then invoked to 'illustrate' (i.e., predict) the underlying polyclonal composition, number of contributing antibodies, and affinity distributions. The claimed quantitative predictions about response structure are therefore the direct output of the fitting procedure rather than an independent derivation or external test.
full rationale
The paper's core argument is that neutralization titer statistics alone enable quantitative predictions of antibody response composition (collective vs. few-dominant) via an equilibrium binding model, with Gumbel distributions arising from extreme-value considerations. However, the model is introduced as one that 'quantitatively captures titer data,' indicating calibration to the same observations used for the structural claims. This reduces the asserted predictive mapping from titers to composition and number/strength distributions to a post-fit interpretation by construction. The Gumbel fit to cohort data is similarly described as 'accurately describe,' reinforcing the pattern. No independent cross-validation (e.g., against sequencing) is indicated in the provided text, but the extreme-value theory component appears external. This constitutes partial circularity under the fitted-input-called-prediction pattern without rendering the entire derivation tautological.
Axiom & Free-Parameter Ledger
Reference graph
Works this paper leans on
-
[1]
kf7VcXpPXsIVYBwamaEYxcW0tlU=
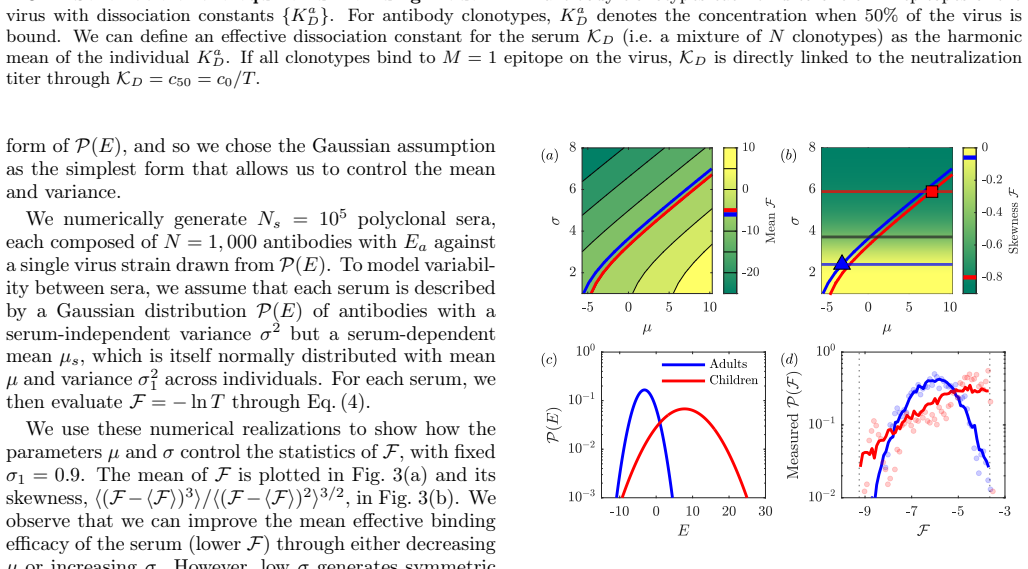
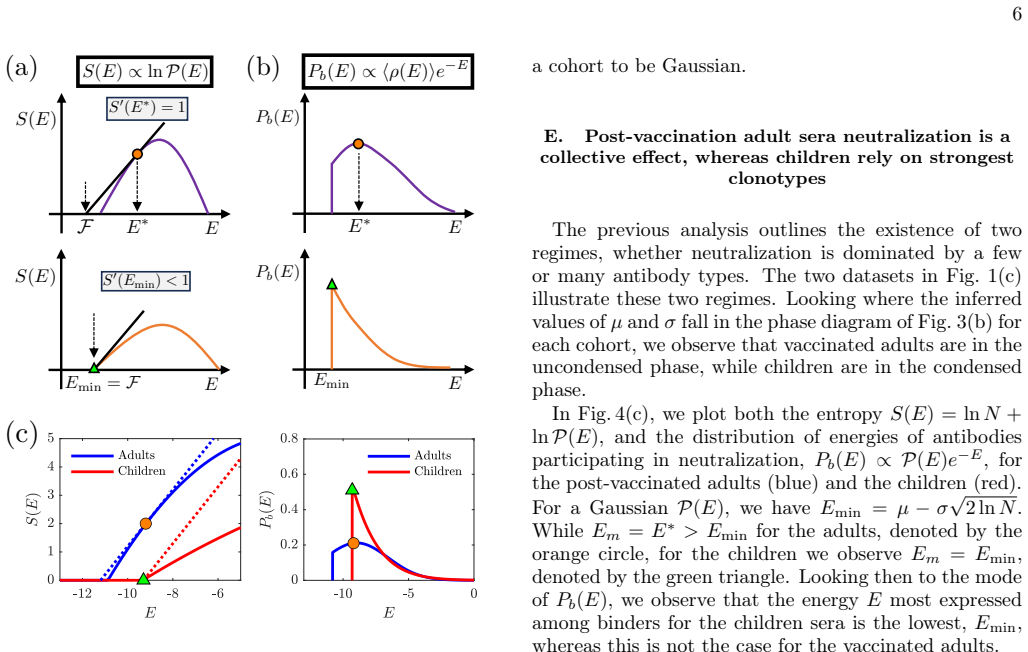
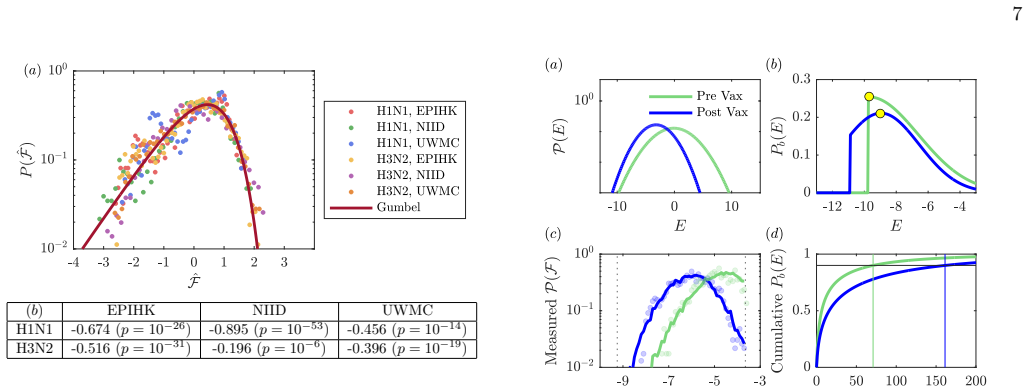
and the United States (University of Washington Medical Center (UWMC),n= 44, ages 21-66) measured against H1N1 and H3N2 flu virus strains from 2024-25. (In analyzing the NIID data, we omit 7 post-vaccination follow up titers.) Separating the data by virus strain (H1N1/H3N2) and by origin of the sera, we identify 6 7 <latexit sha1_base64="kf7VcXpPXsIVYBwam...
2024
-
[2]
bet-hedging
quantifying the change in affinities of B-cells dur- ing affinity maturation suggests that mutations are com- mon (∼0.5/day on average) and some are sizable (35.7% lead to a change in affinity of∆ϕ a >0.7), we choose D= 0.01day −1 as our dynamics describe the change in the average affinity within clonotypeaacross a large number of B-cells trajectories, th...
-
[3]
Jerne NK (1955) The natural-selection theory of anti- body formation.Proceedings of the National Academy of Sciences41:849–857
1955
-
[4]
The Australian Journal of Science20:67–9
Burnet FM (1957) A modification of Jerne’s theory of antibodyproductionusingtheconceptofclonalselection. The Australian Journal of Science20:67–9
1957
-
[5]
Tonegawa S (1983) Somatic generation of antibody di- versity.Nature302:575–581
1983
-
[6]
(1992) Vdj recombination.Immunology Today13:306–314
Alt FW, et al. (1992) Vdj recombination.Immunology Today13:306–314
1992
-
[7]
(2019)Mappingperson-to-personvariation in viral mutations that escape polyclonal serum targeting influenza hemagglutinin.eLife8:e49324
LeeJM,etal. (2019)Mappingperson-to-personvariation in viral mutations that escape polyclonal serum targeting influenza hemagglutinin.eLife8:e49324
2019
-
[8]
Schnaack OH, Nourmohammad A (2021) Optimal evolu- tionary decision-making to store immune memory.eLife 10:e61346
2021
-
[9]
(2021) Comprehensive mapping of mutations in the sars-cov-2 receptor-binding domain that affect recognition by polyclonal human plasma antibod- ies.Cell Host & Microbe29:463–476.e6
Greaney AJ, et al. (2021) Comprehensive mapping of mutations in the sars-cov-2 receptor-binding domain that affect recognition by polyclonal human plasma antibod- ies.Cell Host & Microbe29:463–476.e6. 11
2021
-
[10]
Muñoz-Alía MÁ, Nace RA, Zhang L, Russell SJ (2021) Serotypic evolution of measles virus is constrained by multiple co-dominant b cell epitopes on its surface gly- coproteins.Cell Reports Medicine2:2666–3791
2021
-
[11]
Chardès V, Vergassola M, Walczak AM, Mora T (2022) Affinity maturation for an optimal balance between long- term immune coverage and short-term resource con- straints.Proceedings of the National Academy of Sciences 119:e2113512119
2022
-
[12]
(2020) Deep mutational scanning of sars-cov-2 receptor binding domain reveals constraints on folding and ace2 binding.Cell182:1295–1310.e20
Starr TN, et al. (2020) Deep mutational scanning of sars-cov-2 receptor binding domain reveals constraints on folding and ace2 binding.Cell182:1295–1310.e20
2020
-
[13]
(2021) Complete mapping of mu- tations to the sars-cov-2 spike receptor-binding domain that escape antibody recognition.Cell Host & Microbe 29:44–57.e9
Greaney AJ, et al. (2021) Complete mapping of mu- tations to the sars-cov-2 spike receptor-binding domain that escape antibody recognition.Cell Host & Microbe 29:44–57.e9
2021
-
[14]
(2022) Omicron escapes the majority of ex- istingsars-cov-2neutralizingantibodies.Nature602:657– 663
Cao Y, et al. (2022) Omicron escapes the majority of ex- istingsars-cov-2neutralizingantibodies.Nature602:657– 663
2022
-
[15]
(2022) Ba.2.12.1, ba.4 and ba.5 escape antibodies elicited by omicron infection.Nature608:593– 602
Cao Y, et al. (2022) Ba.2.12.1, ba.4 and ba.5 escape antibodies elicited by omicron infection.Nature608:593– 602
2022
-
[16]
(2009) Broad diversity of neutralizing antibodies isolated from memory b cells in hiv-infected individuals.Nature458:636–640
Scheid JF, et al. (2009) Broad diversity of neutralizing antibodies isolated from memory b cells in hiv-infected individuals.Nature458:636–640
2009
-
[17]
(2022) A biophysical model of viral escape from polyclonal antibodies.Virus Evolution8:veac110
Yu TC, et al. (2022) A biophysical model of viral escape from polyclonal antibodies.Virus Evolution8:veac110
2022
-
[18]
(2013) Molecular deconvolution of the monoclonal antibodies that comprise the polyclonal serum response.Proceedings of the National Academy of Sciences110:2993–2998
Wine Y, et al. (2013) Molecular deconvolution of the monoclonal antibodies that comprise the polyclonal serum response.Proceedings of the National Academy of Sciences110:2993–2998
2013
-
[19]
(2016) Molecular-level analysis of the serum antibody repertoire in young adults before and after sea- sonal influenza vaccination.Nature Medicine22:1456– 1464
Lee J, et al. (2016) Molecular-level analysis of the serum antibody repertoire in young adults before and after sea- sonal influenza vaccination.Nature Medicine22:1456– 1464
2016
-
[20]
(2026) High-throughput neutralization measurements correlate strongly with evolutionary suc- cess of human influenza strains.eLife14:RP106811
Kikawa C, et al. (2026) High-throughput neutralization measurements correlate strongly with evolutionary suc- cess of human influenza strains.eLife14:RP106811
2026
-
[21]
(2025)Nearreal-timedataonthehuman neutralizing antibody landscape to influenza virus to in- form vaccine-strain selection in september 2025.Virus Evolution11:veaf086
KikawaC,etal. (2025)Nearreal-timedataonthehuman neutralizing antibody landscape to influenza virus to in- form vaccine-strain selection in september 2025.Virus Evolution11:veaf086
2025
-
[22]
EinavT,BloomJD (2020)Whentwoarebetterthanone: Modeling the mechanisms of antibody mixtures.PLOS Computational Biology16:1–17
2020
-
[23]
Derrida B (1980) Random-energy model: Limit of a fam- ily of disordered models.Phys. Rev. Lett.45:79–82
1980
-
[24]
Derrida B (1981) Random-energy model: An exactly solvable model of disordered systems.Phys. Rev. B 24:2613–2626
1981
-
[25]
Bryngelson JD, Wolynes PG (1987) Spin glasses and the statistical mechanics of protein folding.Proceedings of the National Academy of Sciences84:7524–7528
1987
-
[26]
Frauenfelder H, Sligar SG, Wolynes PG (1991) The energy landscapes and motions of proteins.Science 254:1598–1603
1991
-
[27]
Bryngelson JD, Onuchic JN, Socci ND, Wolynes PG (1995) Funnels, pathways, and the energy landscape of protein folding: A synthesis.Proteins: Structure, Func- tion, and Bioinformatics21:167–195
1995
-
[28]
Onuchic JN, Luthey-Schulten Z, Wolynes PG (1997) Theory of protein folding: The energy landscape perspec- tive.Annual Review of Physical Chemistry48:545–600
1997
-
[29]
GerlandU,MorozJD,HwaT (2002)Physicalconstraints and functional characteristics of transcription factor–dna interaction.Proceedings of the National Academy of Sci- ences99:12015–12020
2002
-
[30]
cerevisiae.Physical Biology4:134
AurellE,d’HérouëlAF,MalmnäsC,VergassolaM (2007) Transcription factor concentrations versus binding site affinities in the yeast s. cerevisiae.Physical Biology4:134
2007
-
[31]
Mustonen V, Kinney J, Callan CG, Lässig M (2008) Energy-dependent fitness: A quantitative model for the evolution of yeast transcription factor binding sites.Pro- ceedings of the National Academy of Sciences105:12376– 12381
2008
-
[32]
Morán-Tovar R, Lässig M (2024) Nonequilibrium anti- gen recognition during infections and vaccinations.Phys. Rev. X14:031026
2024
-
[33]
Haan L, Ferreira A (2006)Extreme value theory: an introduction(Springer) Vol. 3
2006
-
[34]
Hansen A (2020) The three extreme value distributions: An introductory review.Frontiers in PhysicsVolume 8 - 2020
2020
-
[35]
De Silva NS, Klein U (2015) Dynamics of b cells in ger- minal centres.Nature Reviews Immunology15:137–148
2015
-
[36]
(2016) Visualizing antibody affinity mat- uration in germinal centers.Science351:1048–1054
Tas JMJ, et al. (2016) Visualizing antibody affinity mat- uration in germinal centers.Science351:1048–1054
2016
-
[37]
(2025) Replaying germinal center evo- lution on a quantified affinity landscape.bioRxiv
DeWitt WS, et al. (2025) Replaying germinal center evo- lution on a quantified affinity landscape.bioRxiv
2025
-
[38]
Mayer A, Balasubramanian V, Mora T, Walczak AM (2015) How a well-adapted immune system is orga- nized.Proceedings of the National Academy of Sciences 112:5950–5955
2015
-
[39]
Mayer A, Mora T, Rivoire O, Walczak AM (2016) Di- versity of immune strategies explained by adaptation to pathogen statistics.Proceedings of the National Academy of Sciences113:8630–8635
2016
-
[40]
Lässig M, Mustonen V, Walczak AM (2017) Predicting evolution.Nature Ecology & Evolution1:0077
2017
-
[41]
Lässig M, Mustonen V (2020) Eco-evolutionary control of pathogens.Proceedings of the National Academy of Sciences117:19694–19704
2020
-
[42]
Marchi J, Lässig M, Walczak AM, Mora T (2021) Anti- genic waves of virus–immune coevolution.Proceedings of the National Academy of Sciences118:e2103398118
2021
-
[43]
Chardès V, Mazzolini A, Mora T, Walczak AM (2023) Evolutionary stability of antigenically escaping viruses.Proceedings of the National Academy of Sciences 120:e2307712120
2023
-
[44]
Annual Review of Immunology40:413–442
Victora GD, Nussenzweig MC (2022) Germinal centers. Annual Review of Immunology40:413–442
2022
-
[45]
Zhang J, Shakhnovich EI (2010) Optimality of mutation and selection in germinal centers.PLOS Computational Biology6:1–9
2010
-
[46]
(2015) Salmonella infection drives promiscuous b cell activation followed by extrafollicular affinity maturation.Immunity43:120–131
Di Niro R, et al. (2015) Salmonella infection drives promiscuous b cell activation followed by extrafollicular affinity maturation.Immunity43:120–131
2015
-
[47]
(2016) Complex antigens drive per- missive clonal selection in germinal centers.Immunity 44:542–552
Kuraoka M, et al. (2016) Complex antigens drive per- missive clonal selection in germinal centers.Immunity 44:542–552
2016
-
[48]
(2023) Continuous germinal center inva- sion contributes to the diversity of the immune response
Hägglöf T, et al. (2023) Continuous germinal center inva- sion contributes to the diversity of the immune response. Cell186:147–161.e15
2023
-
[49]
Immunity56:547–561.e7
Chen ST, Oliveira TY, Gazumyan A, Cipolla M, Nussen- zweig MC (2023) B cell receptor signaling in germinal centers prolongs survival and primes b cells for selection. Immunity56:547–561.e7
2023
-
[50]
Sprumont A, Rodrigues A, McGowan SJ, Bannard C, 12 Bannard O (2023) Germinal centers output clonally di- verse plasma cell populations expressing high- and low- affinity antibodies.Cell186:5486–5499.e13
2023
-
[51]
(2023) Antigen presentation dynamics shape the antibody response to variants like sars-cov- 2 omicron after multiple vaccinations with the original strain.Cell Reports42:112256
Yang L, et al. (2023) Antigen presentation dynamics shape the antibody response to variants like sars-cov- 2 omicron after multiple vaccinations with the original strain.Cell Reports42:112256
2023
-
[52]
Schiepers A, van’t Wout MF, Hobbs A, Mesin L, Victora GD (2024) Opposing effects of pre-existing antibody and memory t cell help on the dynamics of recall germinal centers.Immunity57:1618–1628.e4
2024
-
[53]
(2025) Transient silencing of hypermutation preserves b cell affinity during clonal bursting.Nature 641:486–494
Pae J, et al. (2025) Transient silencing of hypermutation preserves b cell affinity during clonal bursting.Nature 641:486–494
2025
-
[54]
Cowling BJ, et al. (2024) Preliminary findings from the dynamics of the immune responses to repeat influenza vaccination exposures (drive i) study: A randomized con- trolled trial.Clinical Infectious Diseases79:901–909
2024
-
[55]
Beek MV, Nussenzweig MC, Chakraborty AK (2022) Two complementary features of humoral immune mem- ory confer protection against the same or variant anti- gens.Proceedings of the National Academy of Sciences 119:e2205598119
2022
-
[56]
(2026) B cell imprinting in children impairs antibodies to the haemagglutinin stalk.Nature
Sun J, et al. (2026) B cell imprinting in children impairs antibodies to the haemagglutinin stalk.Nature
2026
-
[57]
Cobey S, Hensley SE (2017) Immune history and in- fluenza virus susceptibility.Current Opinion in Virology 22:105–111 Emerging viruses: intraspecies transmission; Viral immunology
2017
-
[58]
Lewnard JA, Cobey S (2018) Immune history and in- fluenza vaccine effectiveness.Vaccines6:2076–393X
2018
-
[59]
(2020) Preexisting immunity shapes distinct antibody landscapes after influenza virus infec- tion and vaccination in humans.Science Translational Medicine12:eabd3601
Dugan HL, et al. (2020) Preexisting immunity shapes distinct antibody landscapes after influenza virus infec- tion and vaccination in humans.Science Translational Medicine12:eabd3601
2020
-
[60]
Batista FD, Neuberger MS (1998) Affinity dependence of the b cell response to antigen: A threshold, a ceiling, and the importance of off-rate.Immunity8:751–759
1998
-
[61]
(2010) Polyreactivity increases the apparent affinity of anti-hiv antibodies by heteroligation
Mouquet H, et al. (2010) Polyreactivity increases the apparent affinity of anti-hiv antibodies by heteroligation. Nature467:591–595
2010
-
[62]
(1985) A generalization of the random energy model which includes correlations between energies.J
Derrida, B. (1985) A generalization of the random energy model which includes correlations between energies.J. Physique Lett.46:401–407. 13 Appendix A: Derivation and numerical analysis of Eq.(2) We explain here the form of Eq. (2) which determines the probability that a viral particle is unbound at antibody concentrationc. We denote this probability bypf...
1985
-
[63]
3GLxWd9CH/ISbQR0SD2OtrIM048=
Single epitope,M= 1 First consider the case of a monoclonal antibody of concentrationcwhich binds to a single epitope of a single viral particle with a dissociation constantK D. In a volumeV, the total concentration of viral particles is1/V. The concentration of free viral particles is [free virus]=pfree/V, and that of bound viral particles is [virus-anti...
-
[64]
(2), we want to consider multiple binding epitopes on the viral particle
Multiple epitopes,M >1 To arrive at Eq. (2), we want to consider multiple binding epitopes on the viral particle. We denote byMthe number of epitopes. We make two simplifying assumptions: (i) each antibody clonotype only binds to one epitope on the virus and (ii) binding at distinct epitopes is independent. We defineea as the epitope which clonotypeabinds...
-
[65]
3GLxWd9CH/ISbQR0SD2OtrIM048=
Validating Eqs.(3)and(4)for homogeneous sera We investigate the relation betweenc50 andK D in Eq. (A8) through numerical analysis. We consider a log-normal distribution for the dissociation constants{K a D}(or equivalently GaussianP(ϕ)forϕ a ∝logK a D). We sample N= 1,000values for dissociation constants, repeating thisN s = 104 times. As in the main text...
-
[66]
We perform the same numerical analysis as above, but now withM= 10fixed
Validating Eqs.(3)and(4)for inhomogeneous sera We now show the effect of different serum fractionsfa ̸= 1/Nbetween clonotypes. We perform the same numerical analysis as above, but now withM= 10fixed. We assign each of theNclonotypes a weightw a from a log-normal distribution such thatln(wa)is distributed like a Gaussian with zero mean and standard deviati...
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.