Recognition: unknown
PUFFIN: Protein Unit Discovery with Functional Supervision
Pith reviewed 2026-05-10 09:02 UTC · model grok-4.3
The pith
A graph neural network partitions protein structures into multi-residue units guided by functional annotations.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
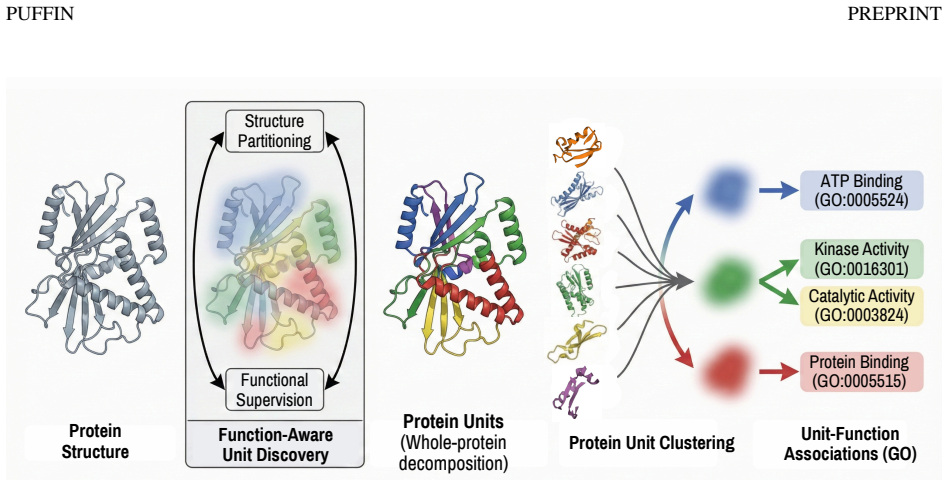
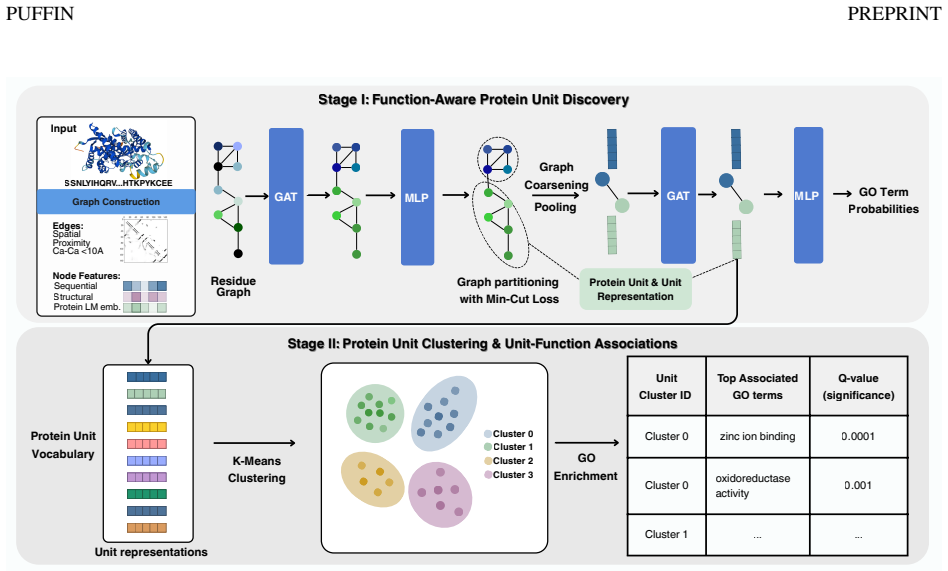
PUFFIN represents proteins as residue-level structure graphs and applies a graph neural network with a structure-aware pooling mechanism that partitions each protein into multi-residue units, with functional supervision that shapes the partition. The learned units are structurally coherent, exhibit organized associations with molecular function, and show meaningful correspondence with curated InterPro annotations. Together these results demonstrate that PUFFIN provides an interpretable framework for analyzing structure-function relationships using learned protein units and their statistical function associations.
What carries the argument
A graph neural network equipped with structure-aware pooling that jointly learns to partition residue graphs into units while incorporating functional supervision to guide the partitions.
If this is right
- The units supply an intermediate-scale view for tracing how coordinated residues enable specific protein activities.
- Statistical associations can be computed between particular units and molecular functions to support targeted interpretation.
- The partitions offer a data-driven alternative or complement to hand-curated domain databases such as InterPro.
- The approach yields units that remain consistent in structure while carrying functional meaning across different proteins.
Where Pith is reading between the lines
- These units could serve as modular targets for protein engineering experiments that alter one function while preserving others.
- Once trained, the model might assign units to unannotated proteins and thereby suggest possible functions without new experiments.
- Combining the units with large-scale structure prediction data could test whether functional groups emerge at consistent stages of folding.
Load-bearing premise
That functional signals from existing annotations are accurate enough to produce partitions reflecting real biological units instead of annotation patterns or data quirks.
What would settle it
Finding that the learned units fail to align better than random groupings with experimental maps of functional residues or with performance on independent function-prediction benchmarks.
Figures

read the original abstract
Proteins carry out biological functions through the coordinated action of groups of residues organized into structural arrangements. These arrangements, which we refer to as protein units, exist at an intermediate scale, being larger than individual residues yet smaller than entire proteins. A deeper understanding of protein function can be achieved by identifying these units and their associations with function. However, existing approaches either focus on residue-level signals, rely on curated annotations, or segment protein structures without incorporating functional information, thereby limiting interpretable analysis of structure-function relationships. We introduce PUFFIN, a data-driven framework for discovering protein units by jointly learning structural partitioning and functional supervision. PUFFIN represents proteins as residue-level structure graphs and applies a graph neural network with a structure-aware pooling mechanism that partitions each protein into multi-residue units, with functional supervision that shapes the partition. We show that the learned units are structurally coherent, exhibit organized associations with molecular function, and show meaningful correspondence with curated InterPro annotations. Together, these results demonstrate that PUFFIN provides an interpretable framework for analyzing structure-function relationships using learned protein units and their statistical function associations. We made our source code available at https://github.com/boun-tabi-lifelu/puffin.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The paper introduces PUFFIN, a graph neural network framework that represents proteins as residue-level structure graphs and applies structure-aware pooling with functional supervision (drawn from annotations such as InterPro and GO) to partition each protein into multi-residue units. The central claim is that the resulting units are structurally coherent, exhibit organized statistical associations with molecular function, and show meaningful correspondence with curated InterPro annotations, thereby providing an interpretable framework for analyzing structure-function relationships.
Significance. If the units can be shown to reflect genuine intermediate-scale biological organization rather than supervision artifacts, the work would offer a novel supervised decomposition approach that integrates structural and functional signals, potentially improving upon purely structural segmentation methods or annotation-dependent analyses. The public release of source code at the cited GitHub repository is a clear strength for reproducibility and further testing.
major comments (2)
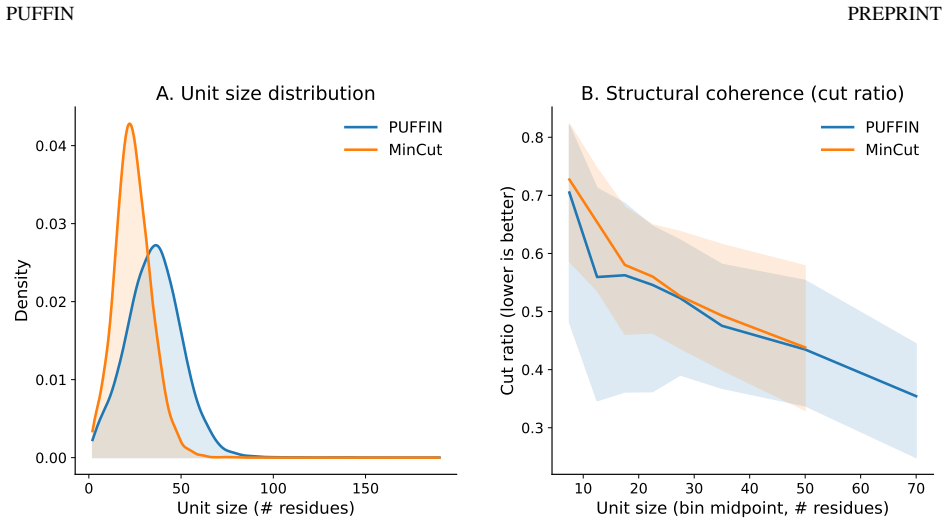
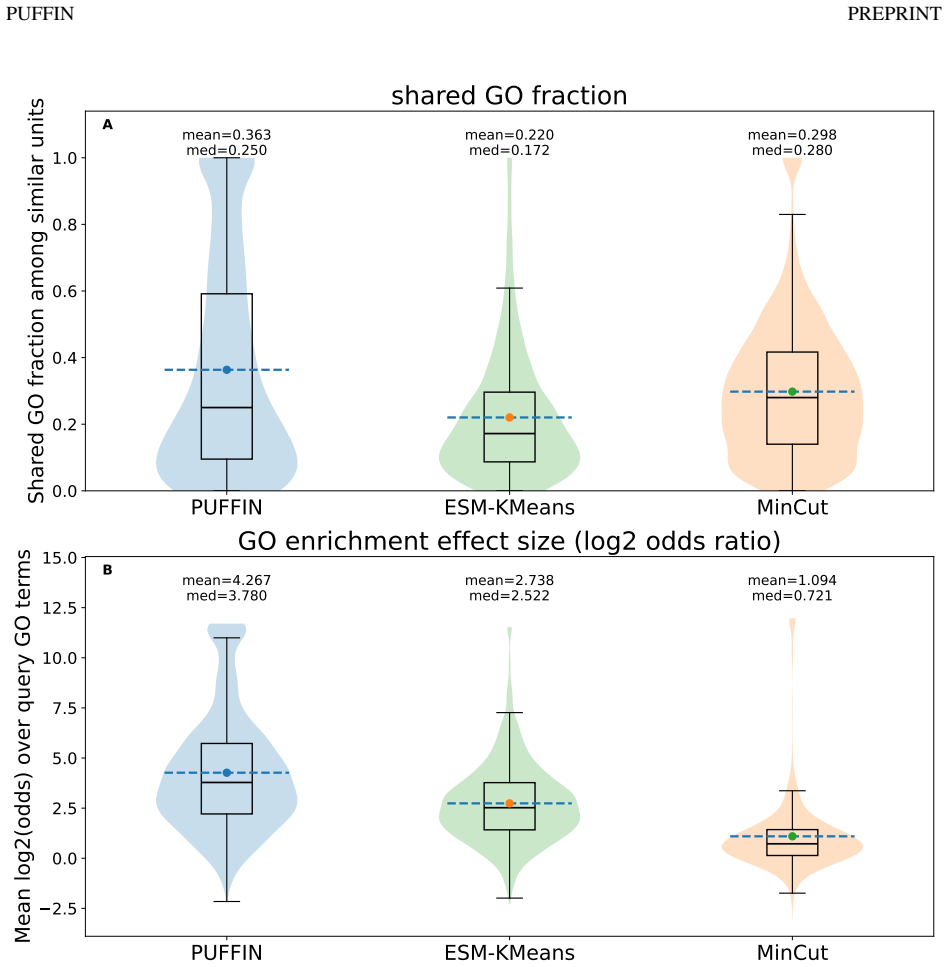
- [Abstract and §4] Abstract and §4 (Results): the claims that learned units are 'structurally coherent,' 'exhibit organized associations with molecular function,' and 'show meaningful correspondence with curated InterPro annotations' are presented without any quantitative metrics, baseline comparisons (e.g., against unsupervised pooling or random partitions), ablation results on the supervision term, or statistical validation details. This absence prevents assessment of whether the data support the central claim that the partitions reflect independent biological units.
- [§3] §3 (Methods, pooling loss): because functional supervision is drawn from the same class of existing annotations later used for InterPro correspondence, the framework lacks an independent test that units would emerge or associate with function under purely structural criteria. If the supervision term dominates the loss, reported coherence and overlap could be artifacts of annotation density and label noise rather than evidence of a new decomposition; an ablation removing or holding out the supervision signal is required to address this.
minor comments (2)
- [Abstract and Introduction] The abstract and introduction should explicitly define the scale and criteria for 'protein units' (e.g., residue count range, structural vs. functional criteria) to avoid ambiguity with existing concepts such as domains or motifs.
- [§2 and §3] Figure captions and method descriptions should clarify the exact graph construction (node/edge features) and the mathematical form of the structure-aware pooling operation for reproducibility.
Simulated Author's Rebuttal
We thank the referee for the constructive comments, which identify key areas where additional evidence and controls would strengthen the manuscript. We address each major comment below and will revise the paper to incorporate quantitative analyses, baselines, and ablations as requested.
read point-by-point responses
-
Referee: [Abstract and §4] Abstract and §4 (Results): the claims that learned units are 'structurally coherent,' 'exhibit organized associations with molecular function,' and 'show meaningful correspondence with curated InterPro annotations' are presented without any quantitative metrics, baseline comparisons (e.g., against unsupervised pooling or random partitions), ablation results on the supervision term, or statistical validation details. This absence prevents assessment of whether the data support the central claim that the partitions reflect independent biological units.
Authors: We agree that the current presentation relies primarily on qualitative descriptions and visualizations. In the revised manuscript we will expand §4 with explicit quantitative metrics (e.g., intra-unit contact density, average residue RMSD within units, and graph modularity), direct comparisons to unsupervised pooling baselines (DiffPool, MinCutPool) and random partitions, an ablation removing the functional supervision term, and statistical validation (permutation tests and hypergeometric enrichment p-values for functional associations and InterPro overlap). The abstract will be updated to reference these quantitative results. These additions will enable readers to evaluate whether the partitions reflect biologically meaningful units. revision: yes
-
Referee: [§3] §3 (Methods, pooling loss): because functional supervision is drawn from the same class of existing annotations later used for InterPro correspondence, the framework lacks an independent test that units would emerge or associate with function under purely structural criteria. If the supervision term dominates the loss, reported coherence and overlap could be artifacts of annotation density and label noise rather than evidence of a new decomposition; an ablation removing or holding out the supervision signal is required to address this.
Authors: We acknowledge the concern that supervision and evaluation draw from related annotation resources. To address this directly, the revised Methods and Results will include an ablation in which the functional supervision loss is removed entirely (training with only the structural partitioning objective). We will compare the resulting units against the full model on structural coherence, functional association statistics, and InterPro overlap. The text will also clarify the distinction between supervision sources (primarily GO terms) and the held-out InterPro evaluation set. This ablation will quantify whether the reported properties arise from structural signals alone or require the supervision term. revision: yes
Circularity Check
No significant circularity detected
full rationale
The paper introduces a GNN-based framework that jointly optimizes structural partitioning and functional supervision drawn from external annotations (e.g., GO, InterPro). No equations, derivations, or parameter-fitting steps are described that reduce a claimed prediction or unit discovery result to its own inputs by construction. The central claims rest on empirical evaluation against held-out structural coherence metrics and annotation overlap, which are independent benchmarks rather than self-referential. No self-citation chains, uniqueness theorems, or ansatz smuggling appear in the load-bearing steps. The approach is therefore self-contained against external data sources.
Axiom & Free-Parameter Ledger
Reference graph
Works this paper leans on
- [1]
-
[2]
Fast Graph Representation Learning with PyTorch Geometric
M. Fey and J. E. Lenssen. Fast graph representation learning with pytorch geometric.preprint arXiv:1903.02428,
work page internal anchor Pith review arXiv 1903
-
[3]
URL https://febs.onlinelibrary.wiley.com/doi/abs/10.1046/j.1432-1033.2002.03130.x
doi: https://doi.org/10.1046/j.1432-1033.2002.03130.x. URL https://febs.onlinelibrary.wiley.com/doi/abs/10.1046/j.1432-1033.2002.03130.x. D. Kingma and J. Ba. Adam: A method for stochastic optimization. InInternational Conference on Learning Representations (ICLR),
- [4]
-
[5]
Let {em}M m=1, em ∈R H, denote the resulting set of unit embeddings across proteins
13 PUFFIN PREPRINT A Unit Cluster Construction After end-to-end training, we extract embeddings for all active units in the training set. Let {em}M m=1, em ∈R H, denote the resulting set of unit embeddings across proteins. Unit embeddings areℓ 2-normalized and transformed using a debiasing procedure fitted on the training set: e′ m = norm em −µ− RX r=1 ⟨e...
2048
-
[6]
Figure S3 shows how these criteria change as the number of prototypes increases. We identify theKvalue, 1024, that balances coverage, specificity, and robustness of functional annotation. GO coverage stays roughly constant as the number of clusters increases, while enrichment-related measures, especially the fraction of enriched clusters, degrade at large...
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.