0
Compact tokenizer generates plausible proteins from scratch with 10x fewer parameters
Yeti: A compact protein structure tokenizer for reconstruction and multi-modal generation
Yeti achieves top codebook use and token diversity, letting a small multimodal model match larger systems in joint sequence and structure出力.
 full image
full image
abstract click to expand
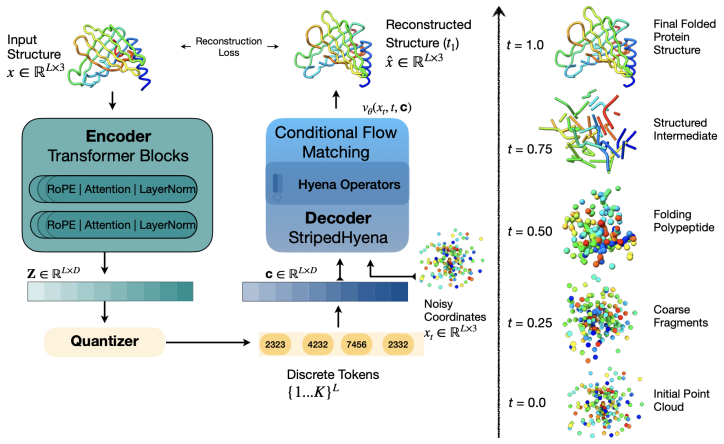
Multimodal models that jointly reason over protein sequences, structures, and function annotations within a unified representation hold immense potential for integrating multimodal data and generating new proteins with designed functional properties. To utilize transformer architectures, such models require a tokenizer that converts protein structure from continuous atomic coordinates into discrete representations suitable for scalable multimodal training. The quality of such models are fundamentally upper bounded by the fidelity and expressiveness of the underlying tokenized structure. However, existing tokenizers prioritize reconstruction over generative abilities. To address these gaps, we introduce Yeti, a simple and compact protein structure tokenizer based on lookup free quantization and trained end to end with a flow matching objective for multimodal learning. Compared to existing models, Yeti generally achieves the best codebook utilization and token diversity, and second best reconstruction accuracy (with 10x fewer parameters than ESM3) on diverse datasets. To validate Yeti's generative capability, we trained a compact multimodal model jointly over its structure tokens and amino acid sequence entirely from scratch, with no pretrained initialization. The resulting multimodal model generates plausible structures under unconditional cogeneration of protein sequence and structures, achieving comparable results to 10x larger models. Together, these results demonstrate that Yeti is a compact and expressive protein structure tokenizer suitable for training multimodal models that cogenerates highly plausible sequences and structures.