Recognition: unknown
Unraveling the Mechanism of Drug Binding to SARS-CoV-2 RNA Pseudoknot with Thermodynamics-Driven Machine Learning
Pith reviewed 2026-05-10 09:59 UTC · model grok-4.3
The pith
Ligand-induced destabilization of the SARS-CoV-2 RNA pseudoknot is selective to its topology and the ligand's protonation state.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
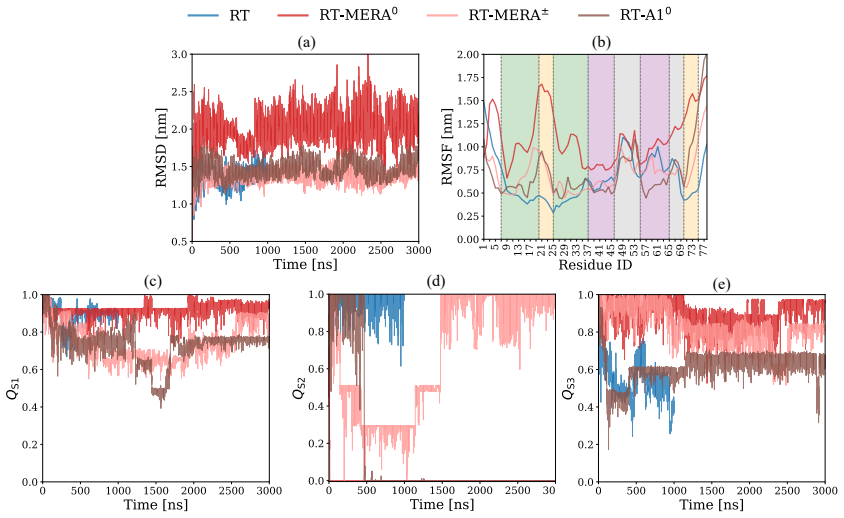
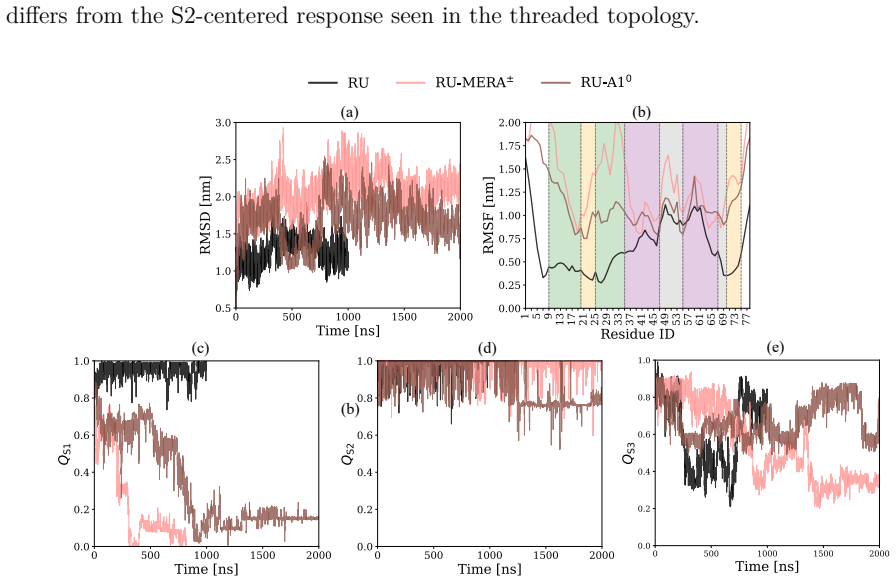
Using spectral map on MD trajectories, the free-energy landscapes demonstrate that merafloxacin and analogs destabilize the S2 stem in the threaded pseudoknot but shift destabilization to S1 and S3 stems in the unthreaded pseudoknot, with the zwitterionic form uniquely imposing slow dynamics on the unthreaded topology, and neutral and zwitterionic forms differing in mechanisms within the same topology.
What carries the argument
Spectral map, a thermodynamics-driven machine learning method that learns collective variables isolating the slowest dynamic modes from MD trajectories of the RNA-ligand complexes.
If this is right
- Physiological protonation states must be considered when modeling RNA-targeted antiviral action.
- Drug design for -1 PRF inhibitors should account for pseudoknot topology to achieve specific destabilization.
- The method can distinguish ligand effects on different RNA conformations.
- Neutral and ionized ligand forms can have qualitatively different mechanisms on the same RNA.
- The unthreaded pseudoknot becomes dynamically active only with certain ligand forms.
Where Pith is reading between the lines
- These findings suggest that targeting specific topologies could improve selectivity of antivirals against SARS-CoV-2.
- Extending the approach to other viral RNAs or ligands might reveal common patterns in frameshifting regulation.
- Experimental validation could involve mutating stems to see if ligand effects change as predicted.
- Protonation-dependent dynamics imply pH could modulate drug efficacy in cellular environments.
Load-bearing premise
The spectral map method correctly identifies the slowest relevant dynamic modes in the MD simulations without being affected by force field inaccuracies or fast fluctuations.
What would settle it
If experiments or higher-level simulations show that the S2 stem remains stable in the threaded form upon ligand binding, or that the zwitterionic form does not slow the unthreaded pseudoknot, the topology-selective destabilization claim would be falsified.
Figures






read the original abstract
The SARS-CoV-2 RNA pseudoknot is a promising target for antiviral intervention, as it regulates the efficiency of $-$1 programmed ribosomal frameshifting ($-$1 PRF), a mechanism that is essential for viral protein synthesis. The pseudoknot represents a viral RNA sequence composed of helical stems that adopts two long-lived topologies, threaded and unthreaded. Ligand-induced distortion of this fold is thought to underlie the susceptibility of $-$1 PRF to small-molecule inhibitors. Resolving these distortions from unbiased molecular dynamics (MD) requires collective variables (CVs) that isolate the slowest dynamic modes of the RNA--ligand system from the high-frequency fluctuations. Here, we use spectral map (SM), a thermodynamics-driven machine-learning method, to learn such CVs directly from MD trajectories of the SARS-CoV-2 RNA pseudoknot in complex with the $-$1 PRF inhibitor merafloxacin and two related analogs. We examine both threaded and unthreaded pseudoknot topologies and consider the neutral and ionized ligand forms relevant at physiological pH. Free-energy landscapes show that ligand-induced destabilization is topology-selective: merafloxacin and its analogs destabilize the S2 stem in the threaded pseudoknot, whereas in the unthreaded pseudoknot, destabilization shifts to the S1 and S3 stems. We find that the zwitterionic form of merafloxacin uniquely imposes slow dynamics on the otherwise featureless unthreaded pseudoknot. Furthermore, the neutral and zwitterionic forms of merafloxacin differ qualitatively in their mechanisms within the same RNA topology. Overall, these results clarify how pseudoknot topology, ligand type, and protonation state shape the slow conformational dynamics of viral RNA and establish physiological protonation as an essential factor for modeling RNA-targeted drug action.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript applies molecular dynamics simulations of the SARS-CoV-2 RNA pseudoknot (threaded and unthreaded topologies) in complex with merafloxacin and two analogs, in both neutral and zwitterionic forms. Spectral map, a thermodynamics-driven machine-learning method, is used to learn collective variables directly from the trajectories. The central claims are that ligand-induced destabilization is topology-selective (S2 stem in threaded pseudoknot; S1/S3 stems in unthreaded), that the zwitterionic form uniquely imposes slow dynamics on the unthreaded pseudoknot, and that neutral versus zwitterionic forms differ qualitatively in mechanism within the same topology.
Significance. If the spectral-map-derived free-energy landscapes are robust, the work would provide concrete mechanistic insight into how small-molecule binding modulates slow conformational dynamics of a viral RNA regulatory element in a manner that depends on both pseudoknot topology and ligand protonation state. This could inform structure-based design of -1 PRF inhibitors. The data-driven extraction of CVs from unbiased MD is a methodological strength when accompanied by appropriate validation.
major comments (2)
- [Methods (spectral map implementation and validation)] The central topology-selective destabilization and zwitterion-specific slow-dynamics claims rest on the spectral-map collective variables accurately isolating ligand-induced slow modes. No convergence diagnostics on the learned eigenvalues, no comparison of the resulting landscapes to those obtained with conventional collective variables (e.g., stem RMSD or base-pair distances), and no repetition with an independent RNA force field are described. These omissions leave open the possibility that the reported shifts in free-energy minima reflect force-field artifacts or incomplete sampling rather than physical ligand effects.
- [Results (free-energy landscapes)] The abstract states that free-energy landscapes were constructed from the SM-projected trajectories, yet no error estimation, block-averaging, or overlap metrics between independent runs are mentioned. Without these, it is difficult to assess whether the observed differences between threaded and unthreaded topologies, or between neutral and zwitterionic ligands, exceed statistical uncertainty.
minor comments (1)
- [Abstract] The abstract would benefit from explicit statement of the total simulation time, number of independent trajectories, and force-field version used, to allow immediate assessment of sampling adequacy.
Simulated Author's Rebuttal
We thank the referee for their constructive and detailed comments, which have prompted us to strengthen the validation aspects of our spectral map analysis. We address each major comment point by point below and have revised the manuscript accordingly to improve clarity and robustness.
read point-by-point responses
-
Referee: [Methods (spectral map implementation and validation)] The central topology-selective destabilization and zwitterion-specific slow-dynamics claims rest on the spectral-map collective variables accurately isolating ligand-induced slow modes. No convergence diagnostics on the learned eigenvalues, no comparison of the resulting landscapes to those obtained with conventional collective variables (e.g., stem RMSD or base-pair distances), and no repetition with an independent RNA force field are described. These omissions leave open the possibility that the reported shifts in free-energy minima reflect force-field artifacts or incomplete sampling rather than physical ligand effects.
Authors: We agree that explicit validation of the spectral map collective variables is essential. In the revised manuscript, we have added convergence diagnostics by monitoring the stability of the learned eigenvalues across successive blocks of each trajectory, demonstrating that the dominant modes converge within the simulation lengths. We have also included side-by-side comparisons of the free-energy landscapes projected onto the spectral map coordinates versus conventional collective variables (stem RMSD and base-pair distances), confirming that the topology-selective minima shifts are reproduced. For the RNA force field, our simulations used the standard Amber ff14SB parameters with RNA-specific modifications; we have expanded the Methods and Discussion sections to justify this choice, cite supporting benchmarks for pseudoknot systems, and explicitly note the absence of independent-force-field repetition as a limitation while arguing that the observed ligand-induced effects are consistent with known RNA-ligand interaction principles. revision: yes
-
Referee: [Results (free-energy landscapes)] The abstract states that free-energy landscapes were constructed from the SM-projected trajectories, yet no error estimation, block-averaging, or overlap metrics between independent runs are mentioned. Without these, it is difficult to assess whether the observed differences between threaded and unthreaded topologies, or between neutral and zwitterionic ligands, exceed statistical uncertainty.
Authors: We thank the referee for highlighting this omission. The revised Results section now incorporates error estimation on all free-energy landscapes via block averaging over non-overlapping trajectory segments, with the resulting uncertainties shown as contour shading. Where multiple independent runs were available for a given system, we report distribution overlap metrics (e.g., Jensen-Shannon divergence) between the projected ensembles. These additions confirm that the reported topology-selective destabilization and zwitterion-specific slow dynamics exceed the estimated statistical uncertainties. revision: yes
Circularity Check
Central results from independent MD trajectories via data-driven spectral map; no load-bearing reduction to fitted parameters or self-citations
full rationale
The derivation proceeds from unbiased MD trajectories of threaded/unthreaded pseudoknots with neutral/zwitterionic ligands, applies spectral map to extract CVs, then constructs free-energy landscapes and identifies topology-selective stem destabilization plus zwitterion-specific slow dynamics. No equation or claim reduces these observations to quantities defined by the method's own fitted parameters or to prior self-citations; the landscapes are direct projections of the sampled trajectories. A minor self-citation for the SM method itself is present but not load-bearing for the reported mechanistic distinctions, which remain falsifiable against the underlying simulation data.
Axiom & Free-Parameter Ledger
axioms (2)
- domain assumption Molecular dynamics simulations with standard force fields sufficiently sample the relevant conformational space of the RNA-ligand complex on the timescales used.
- domain assumption Spectral map correctly identifies collective variables that isolate the slowest modes from high-frequency fluctuations.
Reference graph
Works this paper leans on
-
[1]
(1) Dinman, J. D. Mechanisms and Implications of Programmed Transla- tional Frameshifting.Wiley Interdiscip. Rev. RNA2012,3, 661–673, DOI: https://doi.org/10.1002/wrna.1126. (2) Falese, J. P.; Donlic, A.; Hargrove, A. E. Targeting RNA with Small Molecules: From 27 Fundamental Principles Towards the Clinic.Chem. Soc. Rev.2021,50, 2224–2243, DOI:https://doi...
-
[2]
Schwickert, M.; Hoba, S. N.; Heermann, R.; Kersten, C. Electrostatic Anchoring in RNA-Ligand Design—Dissecting the Effects of Positive Charges on Affinity, Selectivity, Binding Kinetics, and Thermodynamics.J. Med. Chem.2025,68, 8659–8678, DOI: https://doi.org/10.1021/acs.jmedchem.5c00339. (6) Finkel, Y.; Mizrahi, O.; Nachshon, A.; Weingarten-Gabbay, S.; M...
-
[3]
Yahalom-Ronen, Y.; Tamir, H.; Achdout, H.; Stein, D.; Israeli, O.; oth- ers The Coding Capacity of SARS-CoV-2.Nature2021,589, 125–130, DOI: https://doi.org/10.1038/s41586-020-2739-1. (7) Jones, C. P.; Ferré-D’Amaré, A. R. Crystal Structure of the Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) Frameshifting Pseudoknot.RNA2022,28, 239–249, DOI...
-
[4]
Pintilie, G. D.; Rangan, R.; Kladwang, W.; Li, S.; others Cryo-Em and Anti- sense Targeting of the 28-kDa Frameshift Stimulation Element From the SARS- CoV-2 RNA Genome.Nat. Struct. & Mol. Biol.2021,28, 747–754, DOI: https://doi.org/10.1038/s41594-021-00653-y. (10) Bhatt, P. R.; Scaiola, A.; Loughran, G.; Leibundgut, M.; Kratzel, A.; Meurs, R
-
[5]
Dreos, R.; O’Connor, K. M.; McMillan, A.; Bode, J. W.; others Structural Basis of Ribosomal Frameshifting during Translation of the SARS-CoV-2 RNA Genome.Sci- ence2021,372, 1306–1313, DOI:https://doi.org/10.1126/science.abf3546. (11) Peterson, J. M.; Becker, S. T.; O’Leary, C. A.; Juneja, P.; Yang, Y
-
[6]
Moss, W. N. Structure of the SARS-CoV-2 Frameshift Stimulatory Element With an Upstream Multibranch Loop.Biochemistry2024,63, 1287–1296, DOI: https://doi.org/10.1021/acs.biochem.3c00716. (12) Wacker, A.; Weigand, J. E.; Akabayov, S. R.; Altincekic, N.; Bains, J. K
-
[7]
Acids Res.2020,48, 12415–12435, DOI: https://doi.org/10.1093/nar/gkaa1013
Banijamali, E.; Binas, O.; Castillo-Martinez, J.; Cetiner, E.; Ceylan, B.; oth- ers Secondary Structure Determination of Conserved SARS-CoV-2 RNA Ele- ments by NMR Spectroscopy.Nucl. Acids Res.2020,48, 12415–12435, DOI: https://doi.org/10.1093/nar/gkaa1013. (13) Neupane, K.; Zhao, M.; Lyons, A.; Munshi, S.; Ileperuma, S. M.; Ritchie, D. B
-
[8]
Hoffer, N. Q.; Narayan, A.; Woodside, M. T. Structural Dynamics of Single SARS- CoV-2PseudoknotMoleculesRevealTopologicallyDistinctConformers.Nat. Commun. 2021,12, 4749, DOI:https://doi.org/10.1038/s41467-021-25085-6. (14) Pekarek, L.; Zimmer, M. M.; Gribling-Burrer, A.-S.; Buck, S.; Smyth, R
-
[9]
Caliskan, N. Cis-Mediated Interactions of the SARS-CoV-2 Frameshift RNA Alter 29 its Conformations and Affect Function.Nucl. Acids Res.2023,51, 728–743, DOI: https://doi.org/10.1093/nar/gkac1184. (15) Omar, S. I.; Zhao, M.; Sekar, R. V.; Arbabimoghadam, S.; Tuszynski, J. A.; Wood- side, M. T. Modeling the Structure of the Frameshift-Stimulatory Pseudoknot...
-
[10]
Dinman, J. D.; Loerch, S.; Woodside, M. T. Identifying Inhibitors of−1 Programmed Ribosomal Frameshifting in a Broad Spectrum of Coronaviruses.Viruses2022,14, 177, DOI:https://doi.org/10.3390/v14020177. (23) Sun, Y.; Abriola, L.; Niederer, R. O.; Pedersen, S. F.; Alfajaro, M. M.; Silva Mon- teiro, V.; Wilen, C. B.; Ho, Y.-C.; Gilbert, W. V.; Surovtseva, Y...
-
[11]
Rev.2018,118, 4177– 4338, DOI:https://doi.org/10.1021/acs.chemrev.7b00427
Pinamonti, G.; Poblete, S.; Jurecka, P.; others RNA Structural Dynamics as Captured by Molecular Simulations: A Comprehensive Overview.Chem. Rev.2018,118, 4177– 4338, DOI:https://doi.org/10.1021/acs.chemrev.7b00427. 32 (34) Mlynsky, V.; Bussi, G. Exploring RNA Structure and Dynamics Through En- hanced Sampling Simulations.Curr. Opin. Struct. Biol.2018,49,...
-
[12]
Seshadri, S.; Liang, X.; Jones, C. P.; Le Grice, S. F.; Ferre-D’Amare, A. R.; others Discovery of Small Molecules Targeting the Frameshifting Element RNA in SARS-CoV-2 Viral Genome.ACS Med. Chem. Lett.2023,14, 757–765, DOI: https://doi.org/10.1021/acsmedchemlett.3c00051. (49) Trott, O.; Olson, A. J. AutoDock Vina: Improving the Speed and Accuracy of Docki...
-
[13]
Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I.; others CHARMM General Force Field: A Force Field for Drug-like Molecules Compatible with the CHARMM All- Atom Additive Biological Force Fields.J. Comput. Chem.2010,31, 671–690, DOI: https://doi.org/10.1002/jcc.21367. (54) Bussi, G.; Donadio, D.; Parrinello, M. Canonical Sampling Through Velocity Rescaling....
-
[14]
Lin, Z.; Gimelshein, N.; Antiga, L.; others PyTorch: An Imperative Style, High- Performance Deep Learning Library .Adv. Neural Inf. Process. Syst. 33, 8026–8037. (59) Maragliano, L.; Fischer, A.; Vanden-Eijnden, E.; Ciccotti, G. String Method in Col- lective Variables: Minimum Free Energy Paths and Isocommittor Surfaces.J. Chem. Phys.2006,125, 024106, DOI...
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.