Recognition: unknown
FRIGID: Scaling Diffusion-Based Molecular Generation from Mass Spectra at Training and Inference Time
Pith reviewed 2026-05-10 08:34 UTC · model grok-4.3
The pith
FRIGID generates molecular structures from mass spectra with a diffusion language model trained on hundreds of millions of examples and refines outputs at inference time using fragmentation models.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
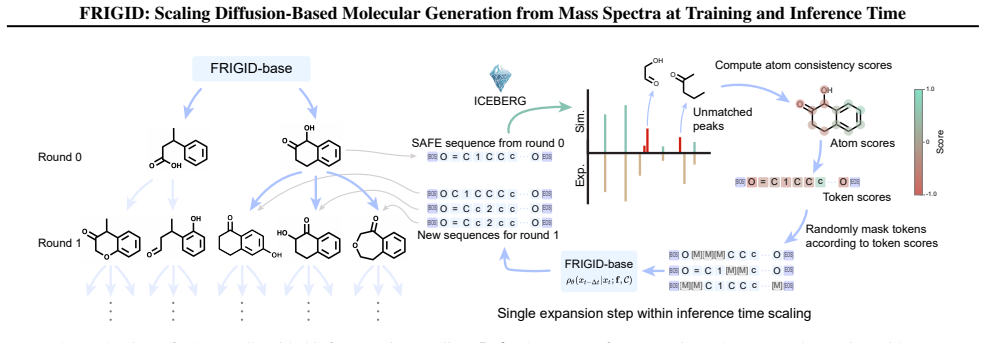
FRIGID is a framework with a novel diffusion language model that generates molecular structures conditioned on mass spectra via intermediate fingerprint representations and determined chemical formulae, training at the scale of hundreds of millions of unlabeled structures. Forward fragmentation models enable inference-time scaling by identifying spectrum-inconsistent fragments and refining them through targeted remasking and denoising, producing significant accuracy gains and log-linear performance scaling with added compute.
What carries the argument
Diffusion language model for spectrum-conditioned molecule generation, paired with forward fragmentation models that drive inference-time refinement via remasking and denoising.
If this is right
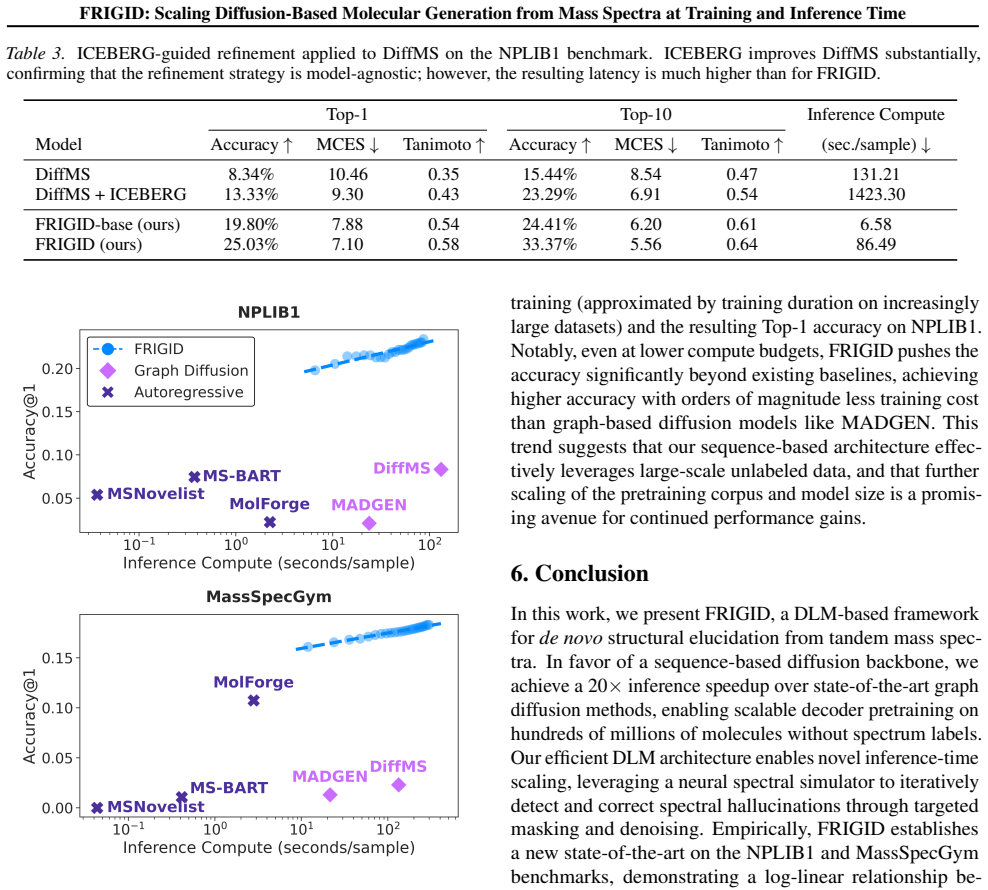
- Surpasses 18 percent top-1 accuracy on the MassSpecGym benchmark.
- Triples the top-1 accuracy of leading prior methods on the NPLIB1 dataset.
- Produces log-linear accuracy gains as inference-time compute is increased.
- Creates a scalable route for continued progress in de novo molecular structure elucidation from spectra.
Where Pith is reading between the lines
- The same refinement loop could be tested on other spectral modalities such as NMR or IR if suitable forward simulators exist.
- Further scaling of the unlabeled training set beyond hundreds of millions could be measured to check whether the log-linear trend continues.
- Real-world deployment would require verifying that the method remains effective when chemical formulae are not known in advance or spectra contain substantial noise.
Load-bearing premise
Forward fragmentation models can accurately and reliably identify spectrum-inconsistent fragments so that targeted remasking and denoising produces genuine improvements rather than new errors.
What would settle it
An experiment on a held-out benchmark set where applying the remasking and denoising step fails to increase the fraction of correct top-ranked structures or reduces overall accuracy.
Figures



read the original abstract
In this work, we present FRIGID, a framework with a novel diffusion language model that generates molecular structures conditioned on mass spectra via intermediate fingerprint representations and determined chemical formulae, training at the scale of hundreds of millions of unlabeled structures. We then demonstrate how forward fragmentation models enable inference-time scaling by identifying spectrum-inconsistent fragments and refining them through targeted remasking and denoising. While FRIGID already achieves strong performance with its diffusion base, inference-time scaling significantly improves its accuracy, surpassing 18% Top-1 accuracy on the challenging MassSpecGym benchmark and tripling the Top-1 accuracy of the leading methods on NPLIB1. Further empirical analyses show that FRIGID exhibits log-linear performance scaling with increasing inference-time compute, opening a promising new direction for continued improvements in de novo structural elucidation. FRIGID code is publicly available at https://github.com/coleygroup/FRIGID
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The paper presents FRIGID, a diffusion language model for de novo molecular structure generation conditioned on mass spectra. It uses intermediate fingerprint representations and determined chemical formulae, with training on hundreds of millions of unlabeled structures. At inference, forward fragmentation models identify spectrum-inconsistent fragments for targeted remasking and denoising, enabling scaling. Reported results include >18% Top-1 accuracy on MassSpecGym, tripling the Top-1 of leading methods on NPLIB1, and log-linear performance gains with increased inference-time compute.
Significance. If the empirical claims hold after proper controls, the work would demonstrate a practical route to scaling molecular generation for mass-spectral elucidation by combining large-scale pretraining with fragmentation-guided refinement. The log-linear scaling observation, if reproducible, would be a notable empirical finding for compute-efficient improvements in this domain.
major comments (2)
- [Abstract] Abstract: the central performance claims (18% Top-1 on MassSpecGym, tripling of leading methods on NPLIB1) are presented without any description of baseline implementations, data splits, leakage controls between training and test spectra, or statistical significance testing; these omissions make it impossible to assess whether the reported gains are load-bearing or artifactual.
- [Inference-time scaling description] The inference-time scaling procedure relies on the forward fragmentation model correctly identifying spectrum-inconsistent fragments; no ablation or error analysis is supplied to show that remasking/denoising produces net gains rather than propagating new errors or overfitting to the refinement loop.
minor comments (1)
- [Abstract] The abstract states that code is publicly available but provides no link or repository details in the main text; this should be added for reproducibility.
Simulated Author's Rebuttal
We thank the referee for their constructive feedback. We address each major comment below and describe the revisions we will incorporate to improve clarity and rigor.
read point-by-point responses
-
Referee: [Abstract] Abstract: the central performance claims (18% Top-1 on MassSpecGym, tripling of leading methods on NPLIB1) are presented without any description of baseline implementations, data splits, leakage controls between training and test spectra, or statistical significance testing; these omissions make it impossible to assess whether the reported gains are load-bearing or artifactual.
Authors: We agree the abstract's brevity omitted key experimental context. The full manuscript details baseline reimplementations following original protocols, use of standard benchmark splits with no spectral leakage (training on separate unlabeled structures), and statistical evaluation via multiple seeds with standard deviations. In revision we will expand the abstract with a concise statement of this evaluation protocol to make the claims more self-contained. revision: yes
-
Referee: [Inference-time scaling description] The inference-time scaling procedure relies on the forward fragmentation model correctly identifying spectrum-inconsistent fragments; no ablation or error analysis is supplied to show that remasking/denoising produces net gains rather than propagating new errors or overfitting to the refinement loop.
Authors: The manuscript reports overall log-linear gains from the full inference procedure and includes empirical scaling curves, but we did not provide a targeted ablation of the fragmentation model's fragment-identification accuracy or an explicit error-propagation analysis. We will add both in the revised manuscript: an ablation comparing performance with and without remasking/denoising, plus quantitative error analysis of the forward model on held-out spectra to confirm net positive contribution. revision: yes
Circularity Check
No significant circularity in derivation chain
full rationale
This is an empirical machine-learning paper that trains a diffusion language model on mass spectra using fingerprint and formula conditioning, then applies forward fragmentation models for inference-time refinement. All performance claims (Top-1 accuracies, log-linear scaling with compute) are presented as measured outcomes on external benchmarks (MassSpecGym, NPLIB1) rather than quantities derived from internal equations or self-referential definitions. No load-bearing step reduces a claimed prediction to a fitted parameter by construction, nor does any uniqueness theorem or ansatz rest solely on prior self-citation. The forward-fragmentation remasking step is treated as an empirical enabler whose effectiveness is validated by ablation-style results, not presupposed. The work is therefore self-contained against external data and does not exhibit the enumerated circularity patterns.
Axiom & Free-Parameter Ledger
Reference graph
Works this paper leans on
- [1]
-
[2]
Bushuiev, R., Bushuiev, A., de Jonge, N
URL https://arxiv.org/ abs/2502.09571. Bushuiev, R., Bushuiev, A., de Jonge, N. F., Young, A., Kretschmer, F., Samusevich, R., Heirman, J., Wang, F., Zhang, L., D ¨uhrkop, K., Ludwig, M., Haupt, N. A., Kalia, A., Brungs, C., Schmid, R., Greiner, R., Wang, B., Wishart, D. S., Liu, L.-P., Rousu, J., Bittremieux, W., Rost, H., Mak, T. D., Hassoun, S., Huber,...
-
[3]
URL https://arxiv.org/abs/2410.23326. Butler, T., Frandsen, A., Lightheart, R., Bargh, B., Taylor, J., Bollerman, T., Kerby, T., West, K., V oronov, G., Moon, K., et al. MS2Mol: A transformer model for illuminating dark chemical space from mass spectra.ChemRxiv,
-
[4]
BERT: Pre-training of deep bidirectional transformers for language understanding
Devlin, J., Chang, M.-W., Lee, K., and Toutanova, K. BERT: Pre-training of deep bidirectional transformers for language understanding. InProceedings of the 2019 Conference of the North American Chapter of the As- sociation for Computational Linguistics: Human Lan- guage Technologies, pp. 4171–4186,
2019
-
[5]
URL https://arxiv. org/abs/2510.20615. Ho, J., Jain, A., and Abbeel, P. Denoising diffusion proba- bilistic models. InProceedings of the 34th International Conference on Neural Information Processing Systems, NIPS ’20, Red Hook, NY , USA,
-
[6]
URLhttps://arxiv.org/abs/2501.06158. Li, X., Thickstun, J., Gulrajani, I., Liang, P. S., and Hashimoto, T. B. Diffusion-lm improves controllable text generation.Advances in Neural Information Process- ing Systems, 35:4328–4343,
-
[7]
and Hutter, F
Loshchilov, I. and Hutter, F. Decoupled weight decay reg- ularization. InProceedings of the Seventh International Conference on Learning Representations (ICLR 2019),
2019
-
[8]
Inference-time scaling for diffusion models beyond scaling denoising steps,
URL https://arxiv.org/abs/ 2501.09732. Manjrekar, M., Bohde, M., Liu, H., Lederbauer, M., Wang, R., and Coley, C. W. Generative structural elucida- tion from mass spectra as an iterative optimization prob- lem,
- [9]
-
[10]
URL https://arxiv.org/abs/ 2310.10773. Prudent, R., Annis, D. A., Dandliker, P. J., Ortholand, J.- Y ., and Roche, D. Exploring new targets and chemical space with affinity selection-mass spectrometry.Nature Reviews Chemistry, 5(1):62–71,
-
[11]
Extended-connectivity fingerprints
doi: 10.1021/ci100050t. Sahoo, S., Arriola, M., Schiff, Y ., Gokaslan, A., Marroquin, E., Chiu, J., Rush, A., and Kuleshov, V . Simple and effective masked diffusion language models.Advances in Neural Information Processing Systems, 37:130136– 130184,
-
[12]
doi: 10.1186/s13321-023-00693-0. URL https://doi. org/10.1186/s13321-023-00693-0. Vaswani, A., Shazeer, N., Parmar, N., Uszkoreit, J., Jones, L., Gomez, A. N., Kaiser, Ł., and Polosukhin, I. Atten- tion is all you need. InNeural Info. Process. Systems, volume 30,
-
[13]
Smiles, a chemical language and information system
doi: 10.1021/ci00057a005. Xing, S., Shen, S., Xu, B., Li, X., and Huan, T. BUDDY: molecular formula discovery via bottom-up MS/MS in- terrogation.Nat. Methods, 20(6):881–890, June
-
[14]
close” and “meaningful
we report additional metrics to further contextualize how close FRIGID predictions are to the true molecule even in cases where the true structure cannot be recovered. In Table 4, we report the percentage of FRIGID samples that correspond to valid molecules as well as the percentage of Top-1 and Top-10 predictions that are “close” and “meaningful” structu...
2023
-
[15]
The best performing model for each metric isboldand the second best is underlined
and MassSpecGym (Bushuiev et al., 2024)de novogeneration datasets. The best performing model for each metric isboldand the second best is underlined. † indicates our implementations of baseline approaches without public code. Methods are approximately ordered by performance. Definitions of meaningful match (Tanimoto similarity of RDKit fingerprints≥0.4 ) ...
2024
-
[16]
Since Neo et al. (2025) did not publicly release their MIST + MolForge implementation, we developed our own reimplementation, and release both the faulty and corrected reimplementations athttps://github.com/harrylaucngd/MIST-MolForge. Table 7.MassSpecGym results for MIST + MolForge under corrected and inflated batched MIST inference. The inflated setting ...
2025
-
[17]
0.812 31.75% 0.68 40.55% 0.74 FRIGID (ours) 0.457 18.29% 0.43 22.00% 0.47 A.4. Robustness to Imperfect Intermediate Predictions The main paper evaluates the standard setting where the chemical formula is assumed known and the molecular fingerprint is predicted from spectra. Here we test robustness when these intermediate signals are imperfect. 14 FRIGID: ...
2026
-
[18]
The full results of this analysis are shown in Table 11, where we find more than 10% of both the NPLIB1 and MassSpecGym test sets overlap with the training set
which we found contains substantial amounts of both NPLIB1 and MassSpecGym test structures. The full results of this analysis are shown in Table 11, where we find more than 10% of both the NPLIB1 and MassSpecGym test sets overlap with the training set. Additionally, the models used to build this simulated spectra dataset were likely trained on datasets wh...
2026
-
[19]
Fingerprint conditioning.We condition on a 4096-bit radius-2 Morgan fingerprint
We do not apply formula conditional dropout during training (pdrop,f =0). Fingerprint conditioning.We condition on a 4096-bit radius-2 Morgan fingerprint. We embed the fingerprint as an unordered set of active bits, retaining up to 256 indices following canonical bit order; in our training configuration, the 16 FRIGID: Scaling Diffusion-Based Molecular Ge...
2023
-
[20]
Our largest model is trained on over 1 billion structures; we found that fine-tuning on NPLIB1 and MassSpecGym did not improve end-to-end performance (Table
with a cosine annealing schedule (Loshchilov & Hutter, 2017). Our largest model is trained on over 1 billion structures; we found that fine-tuning on NPLIB1 and MassSpecGym did not improve end-to-end performance (Table
2017
-
[21]
We train in continuous time (T=0 ) with log-linear noise and antithetic time sampling, using sampling ϵ=10−3
We use mixed precision (bfloat16) and clip gradients to max norm 1.0. We train in continuous time (T=0 ) with log-linear noise and antithetic time sampling, using sampling ϵ=10−3. We maintain an EMA of parameters with decay 0.9999 for evaluation. AdamW hyperparameters are β1=0.9, β2=0.999, ϵ=10−8, and weight decay 0, with peak learning rate3×10 −4. The le...
2014
-
[22]
and MIST + Neuraldecipher (Bohde et al., 2025; Le et al.,
2025
-
[23]
(2025) is not publicly available
as the inference code reproduced in Bohde et al. (2025) is not publicly available. Similarly, to benchmark the GPU hours required to train each baseline in Sec. 5.3, we train each model for one epoch on a single NVIDIA A100 GPU and multiply by the number of epochs used to train each model. B.5. Train Compute Scaling Details In Table 13 we provide settings...
2025
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.