Recognition: unknown
Predictive Modelling of Natural Medicinal Compounds for Alzheimer disease Using Machine Learning and Cheminformatics
Pith reviewed 2026-05-10 02:59 UTC · model grok-4.3
The pith
Machine learning models using molecular descriptors can predict neuroprotective activity of natural compounds for Alzheimer disease.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
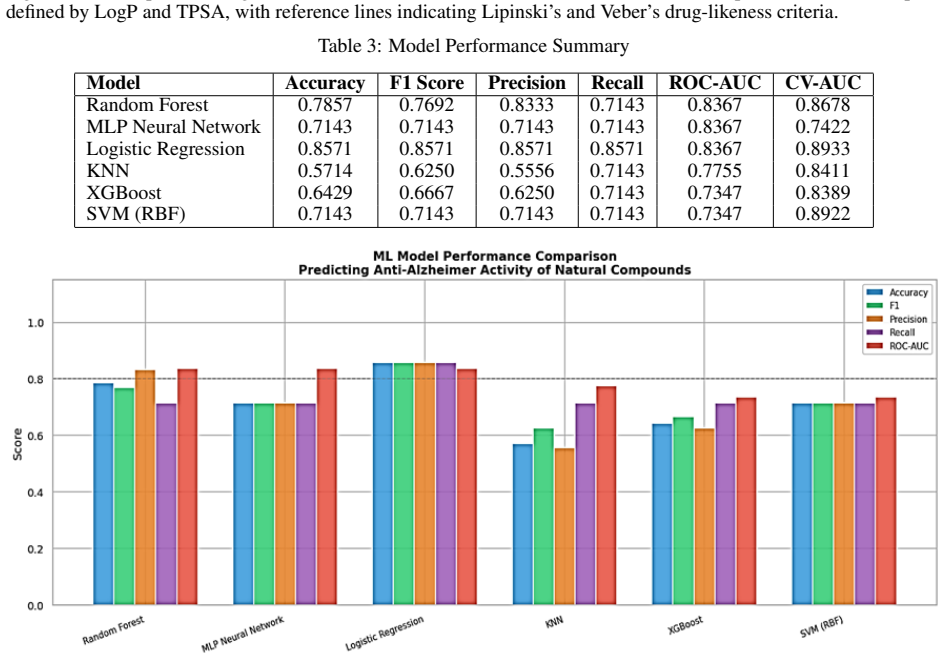
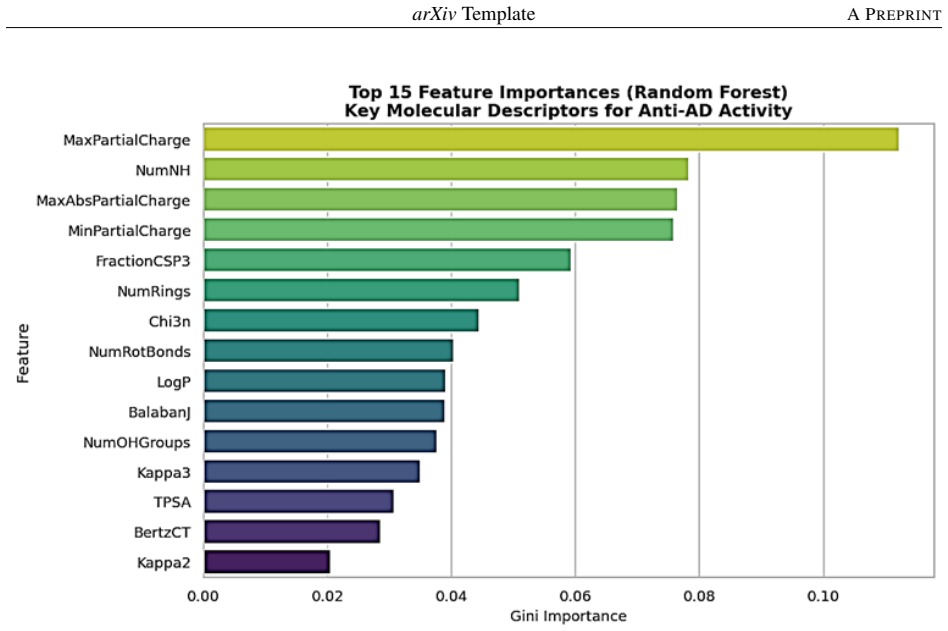
The study shows that Random Forest and similar ensemble classifiers achieve the best accuracy and ROC-AUC when distinguishing active from inactive natural compounds using RDKit-derived physicochemical descriptors. Feature importance analysis identifies lipophilicity, molecular weight, and polarity as the most influential properties for the predicted neuroprotective effects.
What carries the argument
Random Forest ensemble classifier trained on RDKit-computed molecular descriptors (molecular weight, LogP, TPSA, hydrogen-bond counts) from ChEMBL- and PubChem-labeled natural compounds.
If this is right
- Large natural-product libraries can be screened for dementia activity using only computed chemical properties.
- Laboratory resources can be directed first toward compounds the model scores as high-probability actives.
- Lipophilicity and polarity can serve as primary criteria when designing or selecting new neuroprotective candidates.
- The same descriptor-based workflow can be repeated for other neurodegenerative conditions.
Where Pith is reading between the lines
- If the trained model generalizes to compounds outside the original databases, it could cut the volume of initial high-throughput assays required.
- Pairing the classifier with structure-based docking or similarity searches could further narrow the candidate list before synthesis or purchase.
- Public databases become more valuable when used to bootstrap predictive filters rather than only as lookup tables.
Load-bearing premise
Labels for active and inactive compounds taken from public databases correctly represent true anti-dementia biological activity without major errors or bias.
What would settle it
Laboratory testing of a sample of compounds the Random Forest model ranks as highly active, using a standard cell-based or animal model of Alzheimer disease, shows no neuroprotective effect above controls.
Figures




read the original abstract
Alzheimer disease (AD) is a neurodegenerative disease that lacks specific treatment options. Natural drugs have displayed neuroprotective effects; however, their high-throughput discovery is challenging because of the expense of experimental testing.The study proposed a machine learning approach to identify the anti-dementia activity of natural compounds based on molecular descriptors obtained from cheminformatics. The study used a set of active and inactive compounds obtained from public databases like ChEMBL and PubChem. Various molecular descriptors, including molecular weight, lipophilicity (LogP), topological polar surface area (TPSA), and hydrogen bonding descriptors, were calculated with RDKit. Data preprocessing and feature selection were applied, followed by the development of several classification models (Random Forest, XGBoost, Support Vector Machines, Logistic Regression) and their evaluation based on accuracy, precision, recall, F1-score and ROC-AUC. The outcome suggests that ensemble techniques, such as Random Forest, delivered the best predictive accuracy and ROC-AUC values. This study also highlights that critical physicochemical descriptors in particular lipophilicity, molecular weight and polarity are important in driving neuroprotective activity as identified by feature importance analysis. The integrated machine learning approach shows the potential of combining natural product research and machine learning in early drug discovery for dementia. They provide a means of rapidly exploring large datasets and selecting candidates for experimental confirmation, thus minimising costs and time in the development of drugs for neurodegenerative diseases.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript outlines a standard cheminformatics-ML pipeline to classify natural compounds for anti-dementia (neuroprotective) activity. Active and inactive labels are taken from ChEMBL and PubChem; RDKit descriptors (molecular weight, LogP, TPSA, hydrogen-bond counts) are computed; data are preprocessed and features selected; four classifiers (Random Forest, XGBoost, SVM, Logistic Regression) are trained and ranked by accuracy, precision, recall, F1-score and ROC-AUC. The central claims are that Random Forest yields the highest performance and that lipophilicity, molecular weight and polarity are the most important drivers of activity according to feature-importance analysis.
Significance. If the performance numbers and feature rankings survive rigorous validation, the work supplies a practical, low-cost virtual screen for prioritizing natural-product libraries in Alzheimer’s drug discovery. The use of public databases and an ensemble of standard models is a modest but reproducible contribution that could reduce the experimental burden on natural-product screening. Credit is due for the explicit comparison of multiple classifiers and for highlighting physicochemical trends that align with known CNS drug-likeness rules.
major comments (2)
- [Abstract and Methods] Abstract and Methods (Data Acquisition): the binary active/inactive labels are taken directly from ChEMBL and PubChem without any description of the underlying assays, IC50 thresholds, or phenotypic versus target-based criteria. Because the central claim—that Random Forest is superior and that LogP/MW/TPSA drive neuroprotective activity—rests entirely on these labels, the absence of assay-type filtering or confirmation of true negatives is load-bearing. Noisy or assay-specific labels would inflate both accuracy and the reported feature importances.
- [Results] Results section: the abstract asserts that Random Forest delivered the best accuracy and ROC-AUC, yet supplies neither dataset size, active/inactive ratio, cross-validation scheme, nor numerical performance values. Without these quantities it is impossible to judge whether the claimed superiority is statistically meaningful or merely reflects an imbalanced or over-fitted training set.
minor comments (1)
- [Abstract] Abstract: inclusion of at least the final dataset size, the best ROC-AUC value, and the top-three feature-importance ranks would allow readers to assess the claims immediately.
Simulated Author's Rebuttal
We thank the referee for the detailed and constructive feedback, which has helped us identify areas where the manuscript can be strengthened for clarity and scientific rigor. We address each major comment point by point below and will incorporate the necessary revisions into the next version of the manuscript.
read point-by-point responses
-
Referee: [Abstract and Methods] Abstract and Methods (Data Acquisition): the binary active/inactive labels are taken directly from ChEMBL and PubChem without any description of the underlying assays, IC50 thresholds, or phenotypic versus target-based criteria. Because the central claim—that Random Forest is superior and that LogP/MW/TPSA drive neuroprotective activity—rests entirely on these labels, the absence of assay-type filtering or confirmation of true negatives is load-bearing. Noisy or assay-specific labels would inflate both accuracy and the reported feature importances.
Authors: We agree that explicit details on label derivation are essential for validating the central claims. The original manuscript summarized the sources but did not elaborate on assay specifics. In the revised version, we will expand the Data Acquisition section to specify the ChEMBL and PubChem query criteria, including IC50 thresholds for active compounds (e.g., IC50 ≤ 10 μM where available), assay types (target-based vs. phenotypic), and any steps taken to confirm true negatives or filter noisy data. This addition will directly address concerns about label reliability and support the reported feature importances. revision: yes
-
Referee: [Results] Results section: the abstract asserts that Random Forest delivered the best accuracy and ROC-AUC, yet supplies neither dataset size, active/inactive ratio, cross-validation scheme, nor numerical performance values. Without these quantities it is impossible to judge whether the claimed superiority is statistically meaningful or merely reflects an imbalanced or over-fitted training set.
Authors: We acknowledge that the absence of these quantitative details limits the ability to assess model performance rigorously. The current manuscript provides only qualitative statements about Random Forest superiority. In the revision, we will add to the Results section (and update the abstract if space permits) the exact dataset size, active/inactive class ratio, cross-validation procedure (e.g., stratified 5-fold CV with hyperparameter tuning), and all numerical metrics (accuracy, ROC-AUC, precision, recall, F1) for each classifier. We will also include a brief discussion of class imbalance handling and any statistical comparisons to confirm the significance of Random Forest's performance. revision: yes
Circularity Check
No circularity: standard ML training on external database labels
full rationale
The paper applies off-the-shelf classifiers (Random Forest, XGBoost, etc.) to binary activity labels sourced from ChEMBL and PubChem together with RDKit-computed physicochemical descriptors. Performance metrics and feature-importance rankings are obtained by standard train/test splits and cross-validation on those external labels; no internal equation, normalization, or self-citation is used to define the target variable or to force the reported accuracy/ROC-AUC values. The derivation chain is therefore self-contained against independent data sources and does not reduce to any of the enumerated circularity patterns.
Axiom & Free-Parameter Ledger
free parameters (2)
- Model hyperparameters
- Feature selection criteria
axioms (2)
- domain assumption Activity labels from ChEMBL and PubChem accurately reflect true neuroprotective activity without significant noise or bias.
- domain assumption The four classes of RDKit descriptors (MW, LogP, TPSA, H-bond counts) capture the physicochemical features that determine activity.
Reference graph
Works this paper leans on
-
[1]
Alzheimer’s disease drug development pipeline: 2019,
J. Cummings, G. Lee, A. Ritter, M. Sabbagh, and K. Zhong, “Alzheimer’s disease drug development pipeline: 2019,”Alzheimer’s & Dementia: Translational Research & Clinical Interventions, vol. 5, pp. 272–293, 2019
2019
-
[2]
Role of chemoinformatics and machine learning in drug repurposing,
F. Sirci and E. Guney, “Role of chemoinformatics and machine learning in drug repurposing,”Drug Repurposing, vol. 2, no. 1, p. 20250005, 2025. 11 arXivTemplateA PREPRINT
2025
-
[3]
A review of the current status of disease-modifying therapies and prevention of Alzheimer’s disease,
D. V . Parums, “A review of the current status of disease-modifying therapies and prevention of Alzheimer’s disease,”Medical Science Monitor, vol. 30, pp. e945091-1, 2024
2024
-
[4]
The Alzheimer’s disease drug development landscape,
P. Van Bokhovenet al., “The Alzheimer’s disease drug development landscape,”Alzheimer’s Research & Therapy, vol. 13, no. 1, p. 186, 2021
2021
-
[5]
The role of natural product chemistry in drug discovery: Two decades of progress and perspectives,
M. S. Butler and J. J. La Clair, “The role of natural product chemistry in drug discovery: Two decades of progress and perspectives,”Journal of Natural Products, 2025
2025
-
[6]
Simulation-based machine learning approach to classify accelerated biological aging,
S. I. Hasnainet al., “Simulation-based machine learning approach to classify accelerated biological aging,”Sir Syed University Research Journal, vol. 15, no. 2, pp. 23–31, 2025
2025
-
[7]
QSAR-based virtual screening: Advances and applications in drug discovery,
B. J. Neveset al., “QSAR-based virtual screening: Advances and applications in drug discovery,”Frontiers in Pharmacology, vol. 9, p. 1275, 2018
2018
-
[8]
Natural products as sources of new drugs,
D. J. Newman and G. M. Cragg, “Natural products as sources of new drugs,”Journal of Natural Products, vol. 83, no. 3, pp. 770–803, 2020
2020
-
[9]
Molecular descriptors as useful tools,
A. Ion, M. Praisler, and S. Gosav, “Molecular descriptors as useful tools,”Annals of the University of Galati, vol. 44, no. 1, pp. 26–29, 2021
2021
-
[10]
Evaluation of machine learning methods for bipolar disorder detection,
S. I. Hasnainet al., “Evaluation of machine learning methods for bipolar disorder detection,”VF AST Transactions on Software Engineering, vol. 13, no. 3, pp. 129–139, 2025
2025
-
[11]
Natural product for the treatment of Alzheimer’s disease,
T. T. Bui and T. H. Nguyen, “Natural product for the treatment of Alzheimer’s disease,”J. Basic Clin. Physiol. Pharmacol., vol. 28, no. 5, pp. 413–423, 2017
2017
-
[12]
Quantitative structure-activity relationship modeling,
S. C. Peteret al., “Quantitative structure-activity relationship modeling,”Encyclopedia of Bioinformatics, pp. 661–676, 2019
2019
-
[13]
Multi-dimensional QSAR in drug research,
A. Vedani and M. Dobler, “Multi-dimensional QSAR in drug research,”Progress in Drug Research, pp. 105–135, 2000
2000
-
[14]
In silico drug discovery: A machine learning-driven review,
S. Atasever, “In silico drug discovery: A machine learning-driven review,”Medicinal Chemistry Research, vol. 33, no. 9, pp. 1465–1490, 2024
2024
-
[15]
AI and ML driven drug discovery advancements,
D. D. Patelet al., “AI and ML driven drug discovery advancements,”Current Topics in Medicinal Chemistry, 2025
2025
-
[16]
Harnessing machine learning for drug discovery,
A. Husnainet al., “Harnessing machine learning for drug discovery,”Int. J. Multidisciplinary Sciences, vol. 2, no. 4, pp. 149–157, 2023
2023
-
[17]
Prediction of chemical compounds using deep learning,
M. Galushkaet al., “Prediction of chemical compounds using deep learning,”Neural Computing and Applications, vol. 33, no. 20, pp. 13345–13366, 2021
2021
-
[18]
Feature selection for forecasting models,
L. Zhang and J. Wen, “Feature selection for forecasting models,”Energy and Buildings, vol. 183, pp. 428–442, 2019
2019
-
[19]
Building predictive models via feature synthesis,
I. Arnaldo, U.-M. O’Reilly, and K. Veeramachaneni, “Building predictive models via feature synthesis,” inProc. GECCO, 2015, pp. 983–990
2015
-
[20]
Analysis of QSAR research using machine learning,
M. R. Keyvanpour and M. B. Shirzad, “Analysis of QSAR research using machine learning,”Current Drug Discovery Technologies, vol. 18, no. 1, pp. 17–30, 2021
2021
-
[21]
Developing QSAR models using machine learning,
Z. Wang, J. Chen, and H. Hong, “Developing QSAR models using machine learning,”Environmental Science & Technology, vol. 55, no. 10, pp. 6857–6866, 2021
2021
-
[22]
Evolution of QSAR studies with machine learning,
T. A. Soareset al., “Evolution of QSAR studies with machine learning,”ACS Publications, vol. 62, pp. 5317–5320, 2022
2022
-
[23]
Predicting performance using SHAP,
H. Sahlaouiet al., “Predicting performance using SHAP,”IEEE Access, vol. 9, pp. 152688–152703, 2021
2021
-
[24]
Blood–brain barrier challenges,
L. A. Bors and F. Erd ˝o, “Blood–brain barrier challenges,”Scientia Pharmaceutica, vol. 87, no. 1, p. 6, 2019
2019
-
[25]
AI for natural products in neurodegeneration therapies,
F. Fontanellaet al., “AI for natural products in neurodegeneration therapies,”Biomolecules, vol. 16, no. 1, p. 129, 2026
2026
-
[26]
Classification of dopamine receptor ligands using ML,
S. Suprapto and Y . L. Ni’mah, “Classification of dopamine receptor ligands using ML,”Research Journal of Pharmacy and Technology, vol. 17, no. 9, pp. 4507–4514, 2024
2024
-
[27]
AI in drug discovery and clinical relevance,
R. Qureshiet al., “AI in drug discovery and clinical relevance,”Heliyon, vol. 9, no. 7, 2023. 12
2023
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.