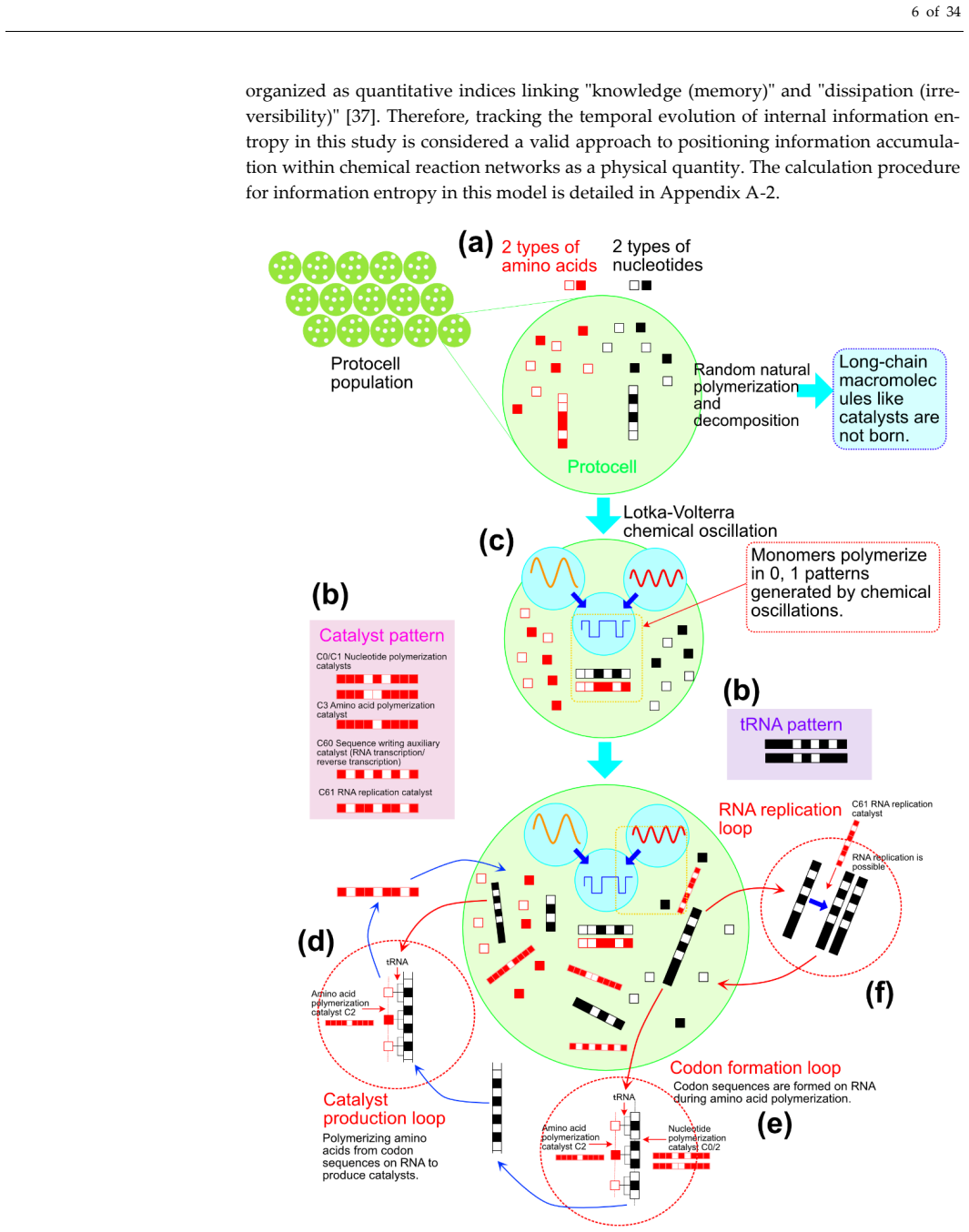
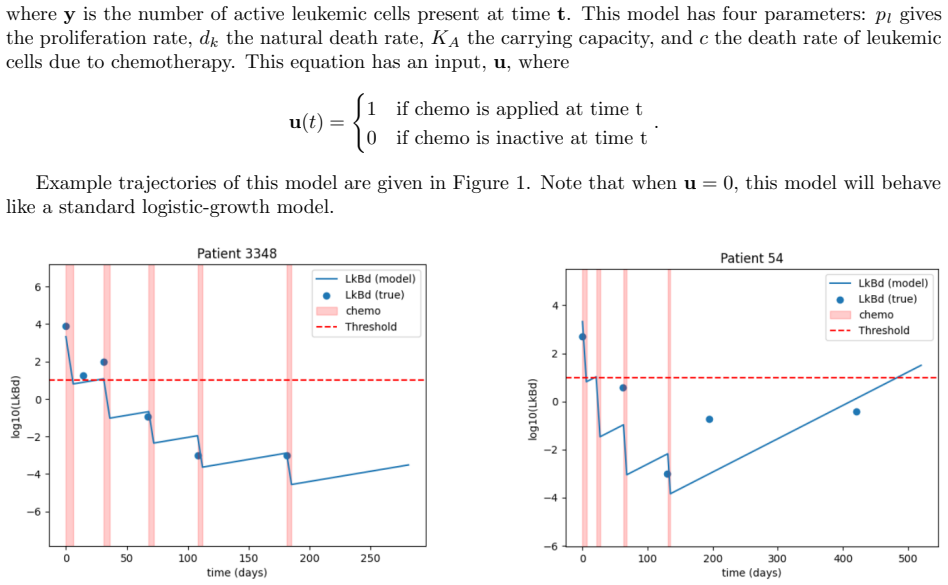
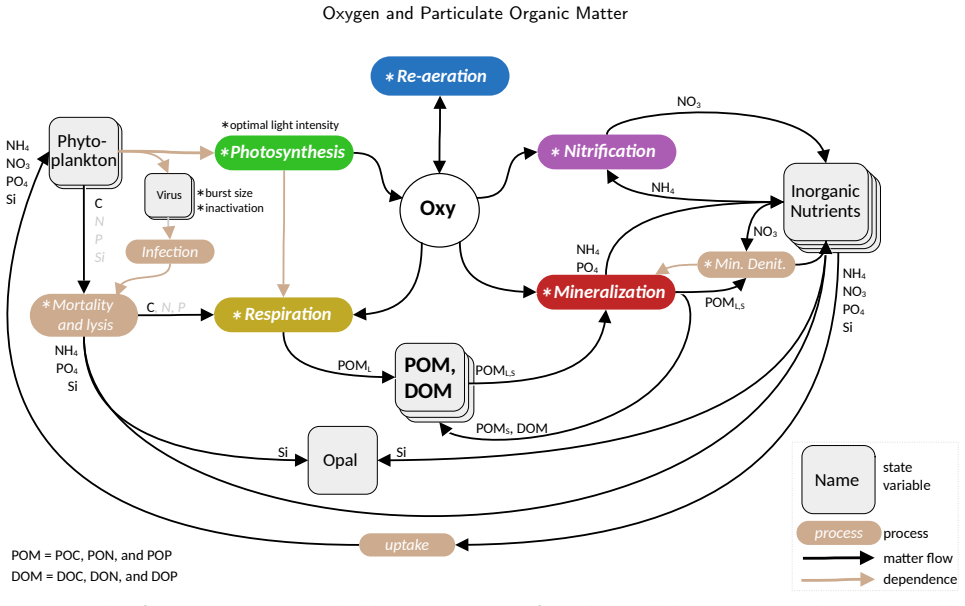
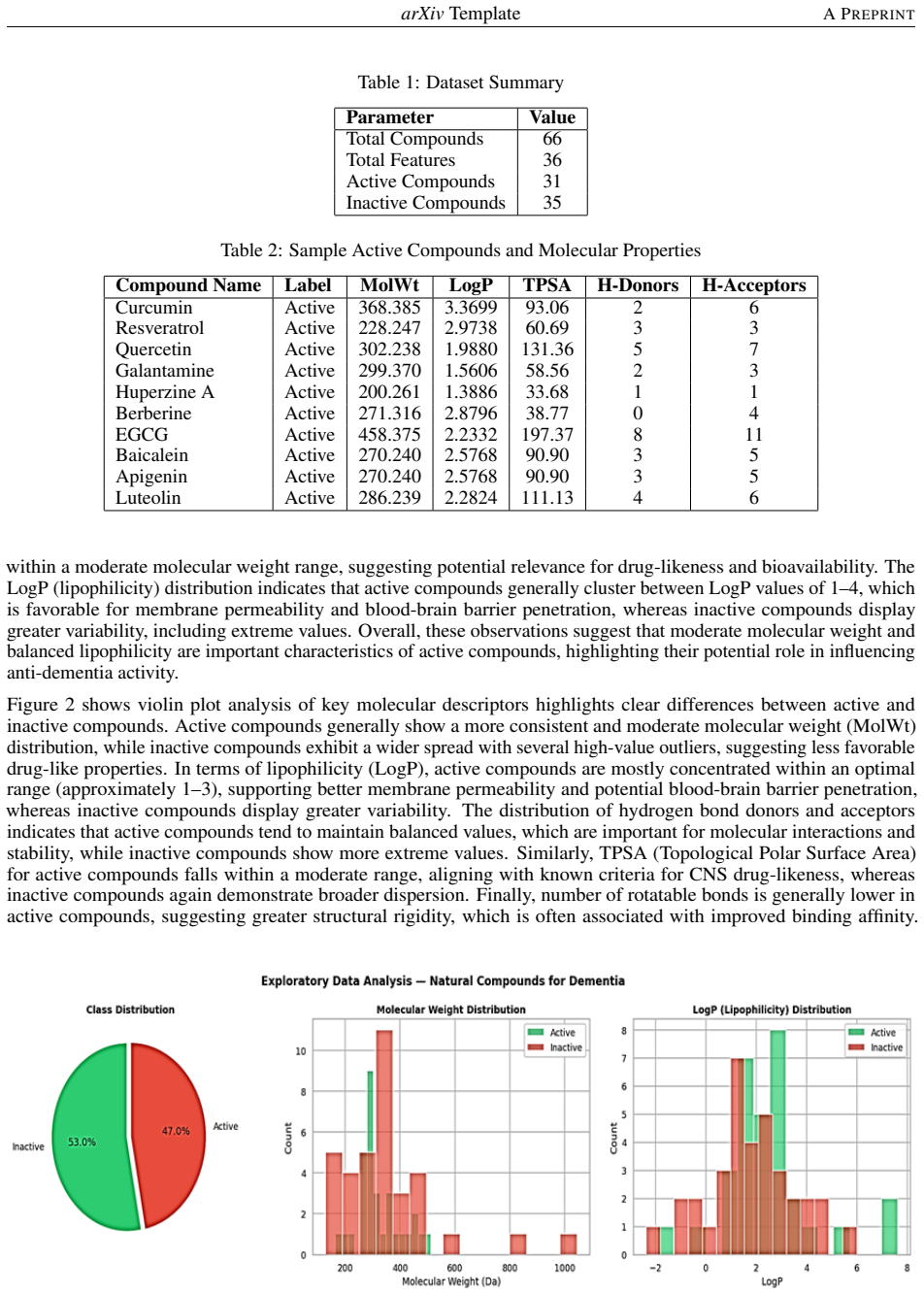
0
Kurdistan study finds seven hawthorn taxa with major fruit variation
Sixty-one accessions vary significantly in weight, size, seeds, pH and moisture, explained by eleven traits.
 full image
full image
abstract click to expand
One of the great phytogeography zones of semi-arid lands in the world is the Kurdistan region of Iraq which hosts many important fruit species due to its geographical location and ecology. Mountain Hawthorn (Crataegus spp.) is a vital wild edible deciduous fruit tree of the genus Crataegus for the region, which is highly beneficial for ornamental, economical, industrial and medicinal uses. In the present study, morphological, phytochemical and molecular marker systems were applied on sixty-one Hawthorn accessions from different locations in the Iraqi Kurdistan region during April 2022 to September 2023. Phenotypic markers have proven to be extremely useful in studies of genetic diversity in Hawthorn genotypes, the results of the present morphological study showed that there are seven taxa (five species, two hybrids) were observed including, Crataegus azarolus, Crataegus meyrei, Crataegus monogyna, Crataegus orientalists, Crataegus pentagyna, Crataegus azarolus x Crataegus meyrei and Crataegus azarolus x Crataegus pentagyna. There was significant variation among different ecotypes in terms of plant type, reproductive stage, and fruit morphology and production uses. Fruit Physio-morphological data revealed a high level of significant variability (P 0.01) among accessions based on the analysis of variance. The most important characteristics for explaining fruit morphological variability `were 11 varbales including fruit weight (FW), fruit length (FL), fruit width (FW), seed length (SL), seed width (SW), number of seeds per fruits (NSF), volume solution (VS), fruit fresh weight (WOF), seed weight (WS), Potentional of hydrogen (pH) and mositure content (MC). They all are significantly different for all the traits measured for the studied accessions.