Recognition: unknown
Quantum AI for Cancer Diagnostic Biomarker Discovery
Pith reviewed 2026-05-10 07:32 UTC · model grok-4.3
The pith
Quantum machine learning applied to multi-omic data identifies combined gene sets as the strongest predictors for distinguishing lung adenocarcinoma from squamous cell carcinoma.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
A quantum classifier trained on the union of differentially expressed and methylated genes outperforms other gene selections for separating LUAD from LUSC tumors and for separating tumors from normal tissue; enrichment analyses link the selected genes to neurotrophin, MAPK, Ras, and PI3K-Akt pathways, supporting their role in subtype-specific oncogenic signaling.
What carries the argument
A quantum machine learning classifier operating on multi-omic gene features, trained after classical differential expression and methylation filtering, with performance evaluated on three sample compositions (LUAD-specific, LUSC-specific, and combined gene sets).
If this is right
- The combined gene set can be used directly as a diagnostic panel for LUAD/LUSC subtyping with reported high predictive metrics.
- Enriched pathways (neurotrophin, MAPK, Ras, PI3K-Akt) become candidate targets for subtype-specific therapeutic follow-up.
- The two-phase workflow (classical feature selection followed by quantum classification) extends to other multi-omic cancer datasets.
- GO and KEGG results supply biological validation that the quantum-selected biomarkers align with known cancer mechanisms.
Where Pith is reading between the lines
- If the quantum advantage holds, similar pipelines could be tested on additional cancer types where multi-omic data volume exceeds classical model capacity.
- The absence of classical baselines leaves open the question of whether the performance gain is due to quantum effects or simply to the chosen gene features.
- The neurotrophin-related genes (NGFR, NTRK2, NTF3) identified here could be validated in independent patient cohorts for prognostic value.
Load-bearing premise
That the quantum classifier supplies a genuine computational advantage over classical methods on the same multi-omic inputs, even though no direct classical baseline comparisons or circuit details are supplied.
What would settle it
A head-to-head accuracy comparison on the identical gene sets using a standard classical random forest or support-vector machine trained and tested under the same cross-validation splits.
Figures













read the original abstract
Quantum machine learning offers a promising new paradigm for computational biology by leveraging quantum mechanical principles to enhance cancer classification, biomarker discovery, and bioinformatics diagnostics. In this study, we apply QML to identify subtype specific biomarkers for lung adenocarcinoma (LUAD) and lung squamous cell carcinoma (LUSC), the two predominant forms of non-small cell lung cancer. Our methodology involves a two-phase process: in Phase 1, differential expression analysis and methylation analysis between tumor and normal samples allows us to identify LUAD-specific and LUSC-specific genes, revealing potential prognostic biomarkers for cancer subtypes. Phase 2 focuses on developing a quantum classifier capable of distinguishing between LUAD and LUSC tumors, as well as between tumor and normal samples. This classifier not only enhances diagnostic precision but also demonstrates the quantum advantage in processing large-scale multiomic datasets. Our results consistently demonstrated that Sample3, representing the combined gene set, achieved the highest overall predictive performance in all metrics. These results demonstrate that QML provides an effective and scalable approach for biomarker discovery and subtype specific cancer classification. GO enrichment analysis highlighted the significant involvement of genes in synaptic signaling, ion channel regulation, and neuronal development. In the quantum phase, KEGG analysis further identified enrichment in cancer-associated pathways, including neurotrophin, MAPK, Ras, and PI3KAkt signaling, with key genes such as NGFR, NTRK2, and NTF3 suggesting a central role in neurotrophinmediated oncogenic processes. Our findings highlight the growing potential of quantum computing to advance precision oncology and next-generation biomedical analytics.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript describes a two-phase approach to cancer biomarker discovery and subtype classification for LUAD and LUSC using quantum machine learning. Phase 1 applies differential expression and methylation analysis to identify subtype-specific genes from multi-omic data. Phase 2 trains a quantum classifier on these gene sets (including a combined 'Sample3' set) to distinguish tumor subtypes and tumor vs. normal samples, reporting highest performance on the combined set along with GO and KEGG enrichments in pathways such as synaptic signaling, MAPK, Ras, and PI3K-Akt.
Significance. If the central claims of measurable quantum advantage, proper validation, and scalability were substantiated with classical baselines and reproducible methods, the work could represent a meaningful step toward practical QML tools in precision oncology and multi-omic analysis. As presented, however, the absence of these elements prevents assessment of whether any performance gains derive from quantum effects or simply from the gene selection process.
major comments (4)
- Abstract and Phase 2: The claim that the quantum classifier 'demonstrates the quantum advantage in processing large-scale multiomic datasets' and that 'QML provides an effective and scalable approach' is unsupported because no quantitative comparisons to classical baselines (SVM, random forest, XGBoost, or kernel methods), no error bars, no statistical tests, and no circuit details (qubit count, ansatz, feature map, optimizer, or loss function) are provided.
- Results (Sample3 performance): The statement that Sample3 'achieved the highest overall predictive performance in all metrics' is reported on gene sets derived from the same data used for training and evaluation, with no mention of held-out test sets, cross-validation procedure, external cohorts, or overfitting controls; this circularity undermines the predictive-performance claim.
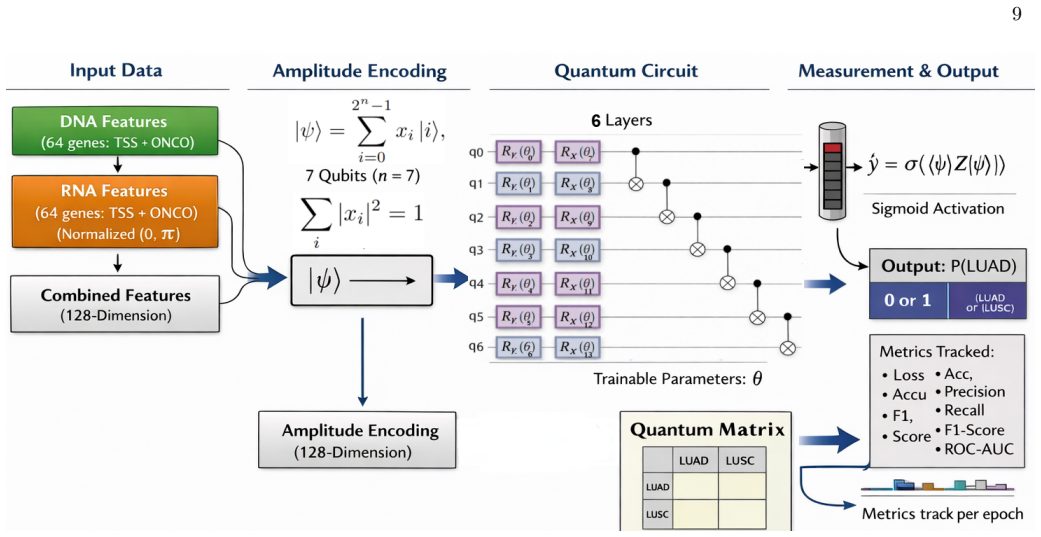
- Methodology (quantum classifier): No description is given of how multi-omic features are encoded into quantum states, the variational circuit architecture, training procedure, or hyperparameter choices, rendering the reported performance metrics impossible to reproduce or attribute to quantum resources rather than classical preprocessing.
- Pathway analysis: The KEGG enrichments (neurotrophin, MAPK, Ras, PI3K-Akt) and highlighted genes (NGFR, NTRK2, NTF3) are presented without statistical thresholds, multiple-testing correction details, or explicit linkage to the quantum classifier outputs, so their relevance to the QML results cannot be evaluated.
minor comments (2)
- The abstract introduces 'Sample3' and 'combined gene set' without a preceding definition or reference to a table/figure that enumerates the gene sets; adding such a table would improve clarity.
- Figure or pseudocode showing the quantum circuit (even if schematic) is absent; its inclusion would aid readers in understanding the claimed quantum component.
Simulated Author's Rebuttal
We thank the referee for their constructive and detailed review. The comments identify important gaps in substantiation, validation, and methodological transparency that we agree must be addressed to strengthen the manuscript. We will undertake a major revision incorporating classical baselines, explicit validation procedures, full quantum circuit specifications, and enhanced statistical details for pathway analysis.
read point-by-point responses
-
Referee: Abstract and Phase 2: The claim that the quantum classifier 'demonstrates the quantum advantage in processing large-scale multiomic datasets' and that 'QML provides an effective and scalable approach' is unsupported because no quantitative comparisons to classical baselines (SVM, random forest, XGBoost, or kernel methods), no error bars, no statistical tests, and no circuit details (qubit count, ansatz, feature map, optimizer, or loss function) are provided.
Authors: We agree that the claims of quantum advantage require direct empirical support. In the revised manuscript we will add a dedicated comparison subsection reporting performance of the quantum classifier against classical baselines (SVM, Random Forest, XGBoost, and kernel methods) trained on identical gene sets. Metrics will be presented with error bars derived from repeated cross-validation, accompanied by statistical tests (e.g., paired t-tests or Wilcoxon tests) for significance. We will also expand the Methods to specify qubit count, feature map, variational ansatz, optimizer, loss function, and all hyperparameter choices. revision: yes
-
Referee: Results (Sample3 performance): The statement that Sample3 'achieved the highest overall predictive performance in all metrics' is reported on gene sets derived from the same data used for training and evaluation, with no mention of held-out test sets, cross-validation procedure, external cohorts, or overfitting controls; this circularity undermines the predictive-performance claim.
Authors: Gene-set selection via differential expression and methylation was performed on the full cohort, which is standard in biomarker discovery. Classifier performance, however, was assessed via k-fold cross-validation. The revised manuscript will explicitly describe the cross-validation protocol (including number of folds and repetition), report metrics with standard deviations, and discuss overfitting safeguards. We acknowledge the lack of an independent external validation cohort and will add this limitation to the Discussion while noting that TCGA data remain the primary benchmark for LUAD/LUSC subtype studies. revision: partial
-
Referee: Methodology (quantum classifier): No description is given of how multi-omic features are encoded into quantum states, the variational circuit architecture, training procedure, or hyperparameter choices, rendering the reported performance metrics impossible to reproduce or attribute to quantum resources rather than classical preprocessing.
Authors: We regret the omission of these essential details. The revised Methods section will provide a complete description of multi-omic feature encoding into quantum states, the variational circuit architecture (ansatz and depth), the training loop (optimizer, iteration count, convergence criteria), and all hyperparameter values together with their selection rationale. This will permit full reproducibility and allow readers to evaluate the contribution of quantum resources. revision: yes
-
Referee: Pathway analysis: The KEGG enrichments (neurotrophin, MAPK, Ras, PI3K-Akt) and highlighted genes (NGFR, NTRK2, NTF3) are presented without statistical thresholds, multiple-testing correction details, or explicit linkage to the quantum classifier outputs, so their relevance to the QML results cannot be evaluated.
Authors: We will revise the pathway analysis to report the exact statistical thresholds employed, the multiple-testing correction method (e.g., Benjamini-Hochberg), and a direct mapping between the enriched pathways/genes and the biomarkers that were subsequently used as input features for the quantum classifier. This will clarify the biological interpretation of the QML results. revision: yes
Circularity Check
No significant circularity in derivation chain
full rationale
The paper presents an empirical two-phase workflow: differential expression and methylation analysis to select subtype-specific genes, followed by training a quantum classifier on those gene sets (including the combined Sample3) and reporting observed performance metrics. No mathematical derivation, first-principles result, or equation is claimed that reduces to its own inputs by construction. The central assertion that Sample3 yields highest performance and that QML is effective/scalable is framed as an outcome of the applied process rather than a self-referential fit or self-citation loop. No load-bearing self-citations, ansatz smuggling, or renaming of known results appear in the provided text. The chain is therefore self-contained as a sequence of data-processing steps.
Axiom & Free-Parameter Ledger
free parameters (1)
- Sample3 gene set selection
axioms (2)
- domain assumption Quantum machine learning models can process large-scale multi-omic datasets more effectively than classical counterparts
- domain assumption Differential expression and methylation differences between tumor and normal samples yield subtype-specific prognostic biomarkers
Reference graph
Works this paper leans on
-
[1]
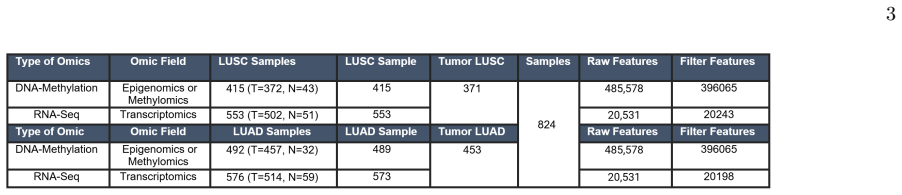
As illustrated in Fig .12, differentially methylated CpGs exhibit distinct patterns across gene regions, CpG island contexts, and chromosomes in LUAD and LUSC
and a multi-omic summary of 22,784 features across 28 dimensions. As illustrated in Fig .12, differentially methylated CpGs exhibit distinct patterns across gene regions, CpG island contexts, and chromosomes in LUAD and LUSC. Performance of Sample 1 Hypermethylated and Downregulated Performance with 64 Features (DN A32 +RN A 32) for Sample 1:The first set...
1977
-
[2]
Board, Non-small cell lung cancer treatment (pdq), PDQ Cancer Information Summaries [Internet] (2024)
P. Board, Non-small cell lung cancer treatment (pdq), PDQ Cancer Information Summaries [Internet] (2024)
2024
- [3]
-
[4]
Biamonte, P
J. Biamonte, P. Wittek, N. Pancotti, P. Rebentrost, N. Wiebe, and S. Lloyd, Quantum machine learning, Na- ture549, 195 (2017)
2017
-
[5]
Xia and S
R. Xia and S. Kais, Quantum machine learning for elec- tronic structure calculations, Nature communications9, 4195 (2018)
2018
-
[6]
Sajjan, S
M. Sajjan, S. H. Sureshbabu, and S. Kais, Quan- tum machine-learning for eigenstate filtration in two- dimensional materials, Journal of the American Chemical Society143, 18426 (2021)
2021
-
[7]
Sajjan, J
M. Sajjan, J. Li, R. Selvarajan, S. H. Sureshbabu, S. S. Kale, R. Gupta, V. Singh, and S. Kais, Quantum machine learning for chemistry and physics, Chemical Society Re- views51, 6475 (2022)
2022
- [8]
-
[9]
Rist` e, M
D. Rist` e, M. P. D. Silva, C. A. Ryan, A. W. Cross, A. D. C´ orcoles, J. A. Smolin, J. M. Gambetta, J. M. Chow, and B. R. Johnson, Demonstration of quantum advan- tage in machine learning, npj Quantum Information3, 16 (2017)
2017
-
[10]
A. S. Bhatia, M. K. Saggi, and S. Kais, Quantum machine learning predicting adme-tox properties in drug discov- ery, Journal of Chemical Information and Modeling63, 6476 (2023)
2023
-
[11]
A. S. Bhatia, S. Kais, and M. A. Alam, Federated quan- volutional neural network: a new paradigm for collabora- 25 tive quantum learning, Quantum Science and Technology 8, 045032 (2023)
2023
-
[12]
A. S. Bhatia, S. Kais, and M. Alam, Handling privacy- sensitive clinical data with federated quantum machine learning, inAPS March Meeting Abstracts, Vol. 2023 (2023) pp. T70–007
2023
-
[13]
Prousalis and N
K. Prousalis and N. Konofaos, A quantum pattern recog- nition method for improving pairwise sequence align- ment, Scientific reports9, 7226 (2019)
2019
-
[14]
A. S. Boev, A. S. Rakitko, S. R. Usmanov, A. N. Kobzeva, I. V. Popov, V. V. Ilinsky, E. O. Kiktenko, and A. K. Fedorov, Genome assembly using quantum and quantum-inspired annealing, Scientific Reports11, 13183 (2021)
2021
-
[15]
E. H. Houssein, Z. Abohashima, M. Elhoseny, and W. M. Mohamed, Hybrid quantum-classical convolutional neu- ral network model for covid-19 prediction using chest x- ray images, Journal of Computational Design and Engi- neering9, 343 (2022)
2022
-
[16]
A. S. Bhatia, M. K. Saggi, A. Kumar, and S. Jain, Matrix product state–based quantum classifier, Neural computa- tion31, 1499 (2019)
2019
-
[17]
M. K. Saggi, A. S. Bhatia, and S. Kais, Federated quan- tum machine learning for drug discovery and healthcare, inAnnual Reports in Computational Chemistry, Vol. 20 (Elsevier, 2024) pp. 269–322
2024
-
[18]
Akpinar and M
E. Akpinar and M. Oduncuoglu, Beyond limits: Chart- ing new horizons in glioma tumor classification through hybrid quantum computing with the cancer genome atlas (tcga) data, in press (2024)
2024
-
[19]
J. W. Chen and J. Dhahbi, Lung adenocarcinoma and lung squamous cell carcinoma cancer classification, biomarker identification, and gene expression analysis us- ing overlapping feature selection methods, Scientific re- ports11, 13323 (2021)
2021
-
[20]
Huang, L
Z. Huang, L. Chen, and C. Wang, Classifying lung ade- nocarcinoma and squamous cell carcinoma using rna-seq data, Cancer Stud Mol Med Open J3, 27 (2017)
2017
-
[21]
L. Xu, Z. Huang, Z. Zeng, J. Li, H. Xie, and C. Xie, An integrative analysis of dna methylation and gene expres- sion to predict lung adenocarcinoma prognosis, Frontiers in Genetics13, 970507 (2022)
2022
-
[22]
Goldman, B
M. Goldman, B. Craft, M. Hastie, K. Repeˇ cka, F. Mc- Dade, A. Kamath, A. Banerjee, Y. Luo, D. Rogers, A. Brooks, and Others, Visualizing and interpreting can- cer genomics data via the xena platform, Nature Biotech- nology38, 675 (2020)
2020
-
[23]
Esteller, Cancer epigenomics: Dna methylomes and histone-modification maps, Nature Reviews Genetics8, 286 (2007)
M. Esteller, Cancer epigenomics: Dna methylomes and histone-modification maps, Nature Reviews Genetics8, 286 (2007)
2007
-
[24]
Network and Others, Comprehensive genomic charac- terization of squamous cell lung cancers, Nature489, 519 (2012)
C. Network and Others, Comprehensive genomic charac- terization of squamous cell lung cancers, Nature489, 519 (2012)
2012
-
[25]
Zhang, I
X. Zhang, I. Jonassen, and A. Goksøyr, Machine learning approaches for biomarker discovery using gene expression data, Exon Publications , 53 (2021)
2021
-
[26]
Chaudhary, O
K. Chaudhary, O. B. Poirion, L. Lu, and L. X. Garmire, Deep learning-based multi-omics integration robustly predicts survival in liver cancer, Clinical Cancer Research 24, 1248 (2018)
2018
-
[27]
Y. Wang, L. Zhang, Y. Liu, and J. Li, Machine learning approaches for biomarker discovery in lung adenocarci- noma using tcga data, Frontiers in Genetics14, 116789 (2023)
2023
-
[28]
Q. Li, Y. Sun, Y. Zhang,et al., Support vector machine- based gene selection for lung cancer subtype classification using tcga rna-seq data, BMC Bioinformatics24, 112 (2023)
2023
-
[29]
R. Chen, J. Li, and Y. Sun, Autoencoder-based feature extraction for biomarker discovery in non-small cell lung cancer, IEEE/ACM Transactions on Computational Bi- ology and Bioinformatics19, 1954 (2022)
1954
-
[30]
Zhang, X
L. Zhang, X. Chen, Y. Li,et al., Hybrid deep learning framework for biomarker identification in lung adenocar- cinoma, BMC Bioinformatics20, 621 (2019)
2019
-
[31]
Huang, J
Z. Huang, J. Yang, and Q. Zhang, Multi-omics integra- tion and machine learning for lung cancer subtype clas- sification and biomarker discovery, Scientific Reports13, 3214 (2023)
2023
- [32]
-
[33]
M. K. Saggi and S. Kais, Mqml: Multi-omic quantum machine learning based cancer classification, biomarker identification in human lung adenocarcinoma, in2024 IEEE International Conference on Quantum Computing and Engineering (QCE), Vol. 1 (IEEE, 2024) pp. 1713– 1720
2024
-
[34]
Relli, M
V. Relli, M. Trerotola, E. Guerra, and S. Alberti, Aban- doning the notion of non-small cell lung cancer, Trends In Molecular Medicine25, 585 (2019)
2019
-
[35]
X. Ji, Y. Boss´ e, M. T. Landi, J. Gui, X. Xiao, D. Qian, P. Joubert, M. Lamontagne, Y. Li, I. Gorlov,et al., Iden- tification of susceptibility pathways for the role of chro- mosome 15q25. 1 in modifying lung cancer risk, Nature Communications9, 3221 (2018)
2018
-
[36]
Peng, C.-C
T.-J. Peng, C.-C. Chang Wang, S.-J. Tang, G.-H. Sun, and K.-H. Sun, Neurotrophin-3 facilitates stemness prop- erties and associates with poor survival in lung cancer, Neuroendocrinology114, 921 (2024)
2024
-
[37]
K. W. Sinkevicius, C. Kriegel, K. J. Bellaria, J. Lee, A. N. Lau, K. T. Leeman, P. Zhou, A. M. Beede, C. M. Fill- more, D. Caswell,et al., Neurotrophin receptor trkb pro- motes lung adenocarcinoma metastasis, Proceedings of the National Academy of Sciences111, 10299 (2014)
2014
-
[38]
Ricci, C
A. Ricci, C. Salvucci, S. Castelli, A. Carraturo, C. De Vi- tis, and M. D’Ascanio, Adenocarcinomas of the lung and neurotrophin system: a review, Biomedicines10, 2531 (2022)
2022
-
[39]
X. Wang, Z. Xu, X. Chen, X. Ren, J. Wei, S. Zhou, X. Yang, S. Zeng, L. Qian, G. Wu,et al., A tropomyosin receptor kinase family protein, ntrk2 is a potential predic- tive biomarker for lung adenocarcinoma, PeerJ7, e7125 (2019)
2019
-
[40]
Palmieri, B
C. Palmieri, B. Rudraraju, M. Monteverde, L. Lattanzio, O. Gojis, R. Brizio, O. Garrone, M. Merlano, N. Syed, C. Lo Nigro,et al., Methylation of the calcium channel regulatory subunitα2δ-3 (cacna2d3) predicts site-specific relapse in oestrogen receptor-positive primary breast car- cinomas, British journal of cancer107, 375 (2012)
2012
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.