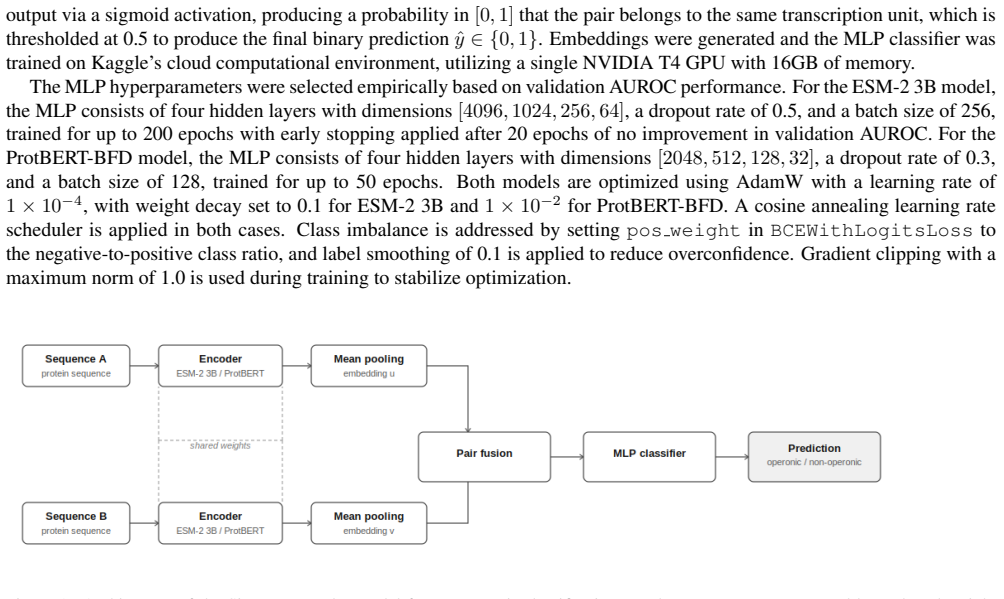
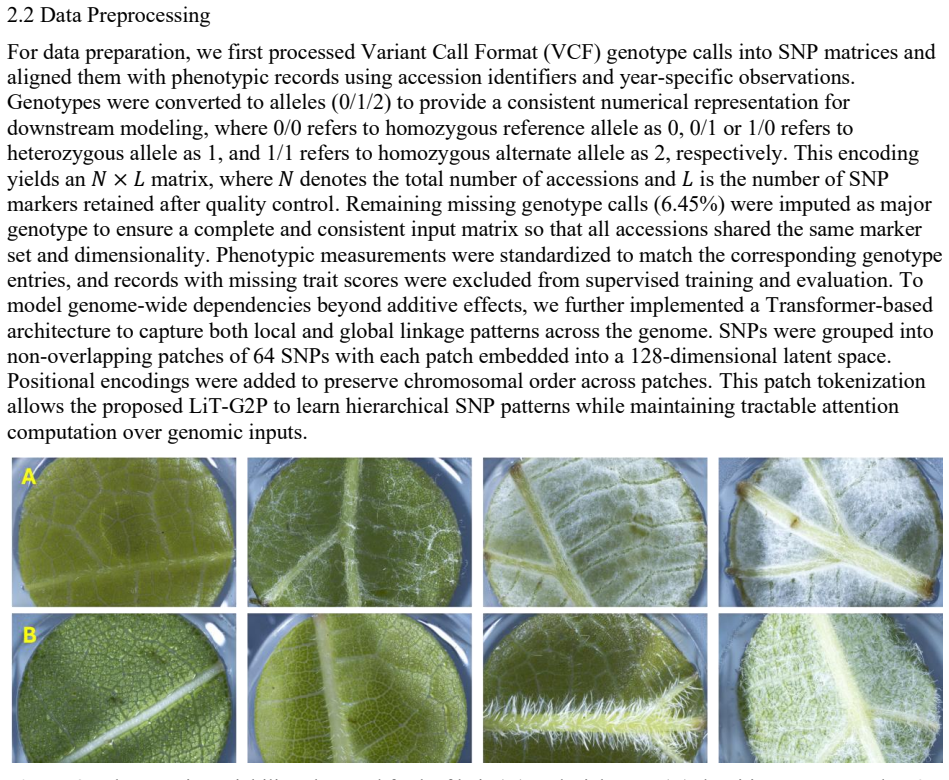
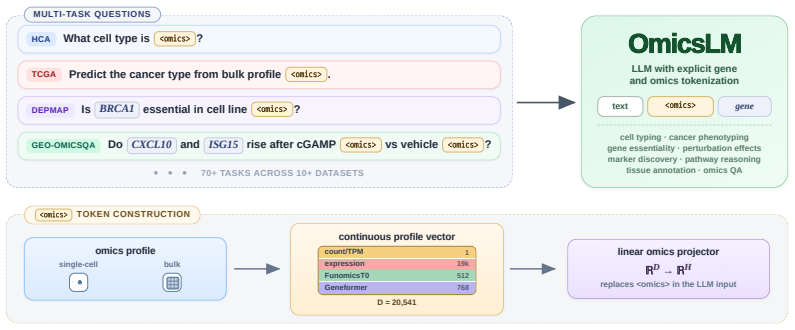
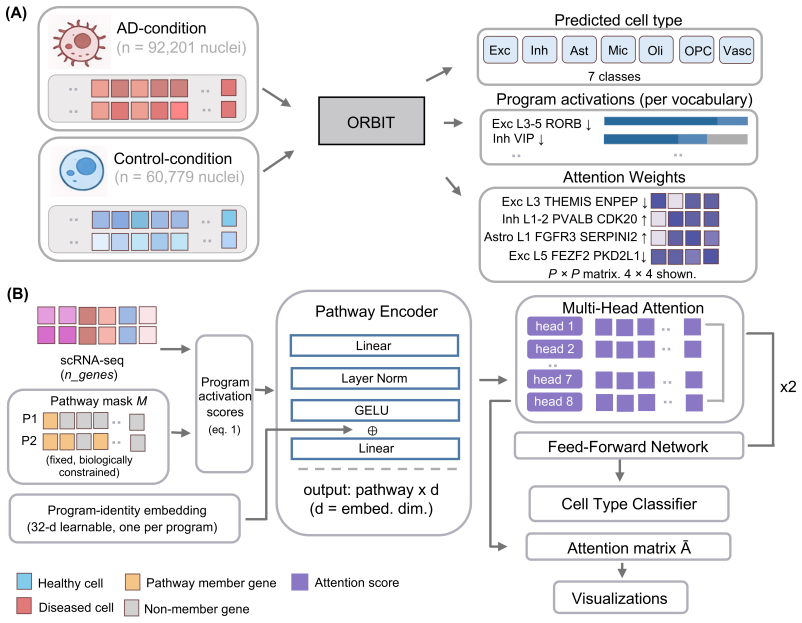
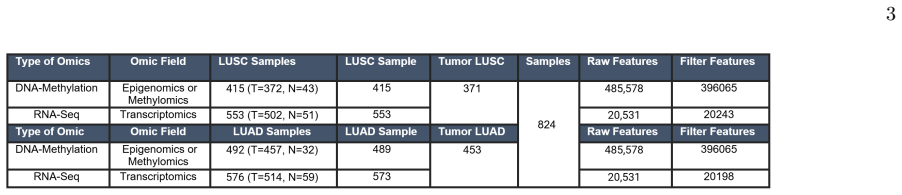
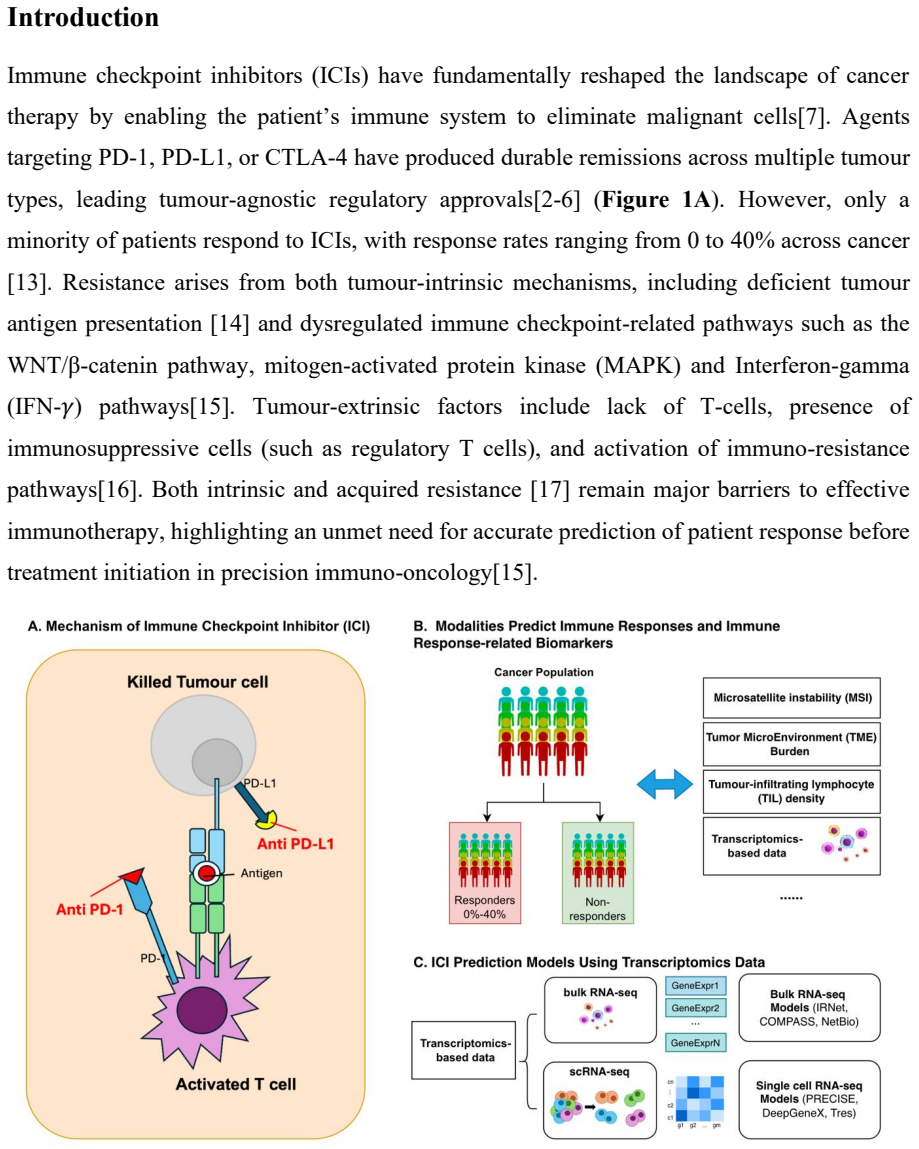
0
Genome embeddings predict microbiome abundances for novel species
Set-Aggregated Genome Embeddings for Microbiome Abundance Prediction
Set-aggregated representations from genomic language models generalize better than classical bioinformatics methods on unseen genomes.
 full image
full image
abstract click to expand
Microbiome functions are encoded within the genes of the community-wide metagenome. A natural question is whether properties of a microbial community can be predicted just from knowing the raw DNA sequences of its members. In this work, we employ set-aggregated genome embeddings (SAGE) to predict community-level abundance profiles, exploiting the few-shot learning capabilities of genomic language models (GLMs). We benchmark this approach to show improved generalization on novel genomes compared to classical bioinformatics approaches. Model ablation shows that community-level latent representations directly result in improved performance. Lastly, we demonstrate the benefits of intermediate transformations between latent representations and demonstrate the differences between GLM embedding choices.