Recognition: unknown
Bayesian inference for disease transmission models informed by viral dynamics
Pith reviewed 2026-05-09 23:43 UTC · model grok-4.3
The pith
A multiscale Bayesian model links viral load trajectories to household transmission parameters via a cut inference approach.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
We propose a multiscale model that jointly captures heterogeneous individual-level viral load trajectories and stochastic household transmission, and develop efficient inference methods to fit it to data. Since full joint inference is computationally difficult, we employ a cut approach that passes information from the within-host to the between-host model but not vice versa. This enables the data on viral loads to inform the transmission parameters such as the infection times and symptom onset thresholds. We evaluate the framework on simulated household outbreak data, assessing parameter recovery, computational efficiency, and the effect of viral load sampling frequency on inference quality.
What carries the argument
The unidirectional cut inference method that feeds within-host viral load information into the between-host transmission model without feedback.
If this is right
- Viral load observations can directly sharpen estimates of infection timing and symptom thresholds in household outbreaks.
- The cut method keeps computation feasible while still letting individual-scale data inform population-scale transmission parameters.
- Sparse viral sampling introduces bias that external viral load datasets can partially correct.
- The framework supports joint analysis of heterogeneous viral trajectories and stochastic transmission events.
Where Pith is reading between the lines
- This structure could support real-time analysis of outbreak data by updating transmission estimates as new viral measurements arrive.
- The same cut technique might transfer to other multiscale disease models where full joint fitting remains intractable.
- Accounting for measurement error in viral loads within the within-host component could further reduce bias under sparse sampling.
Load-bearing premise
The one-way cut from viral dynamics to transmission parameters is enough to keep estimates of infection times and symptom thresholds accurate and unbiased.
What would settle it
A simulation with high-frequency viral load sampling that still produces biased estimates of household infection times would disprove the unbiased recovery result.
Figures
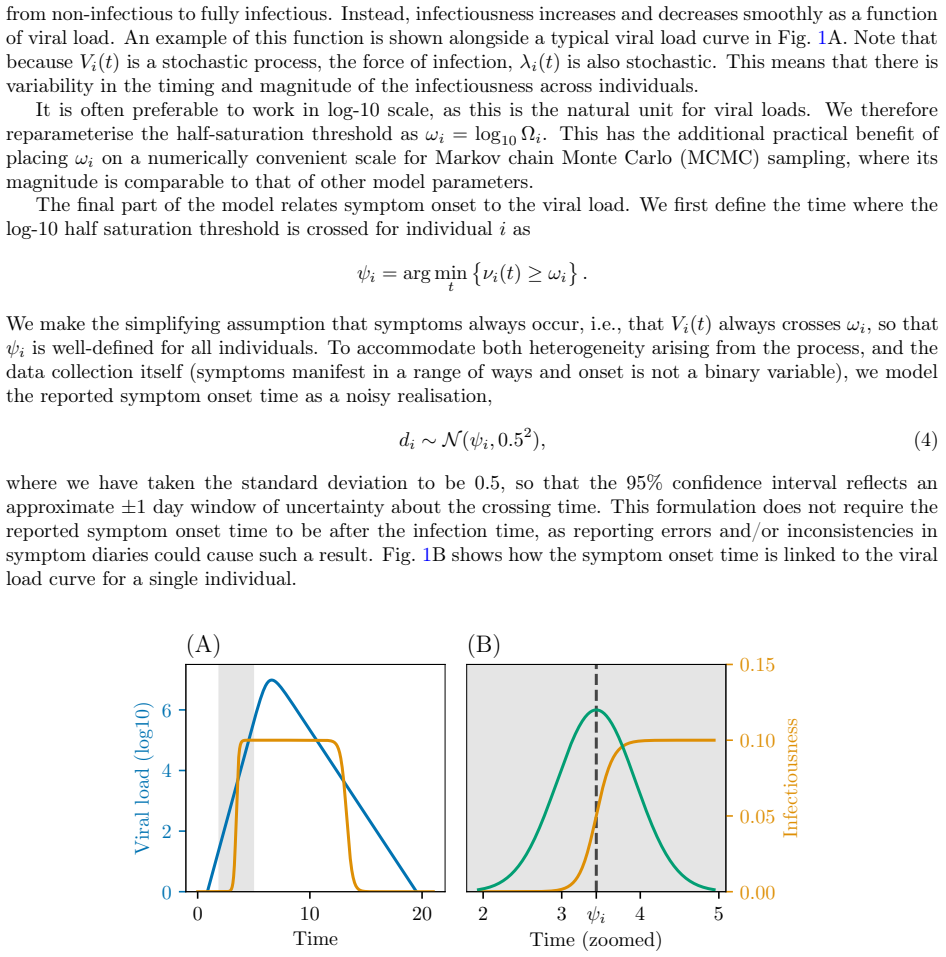


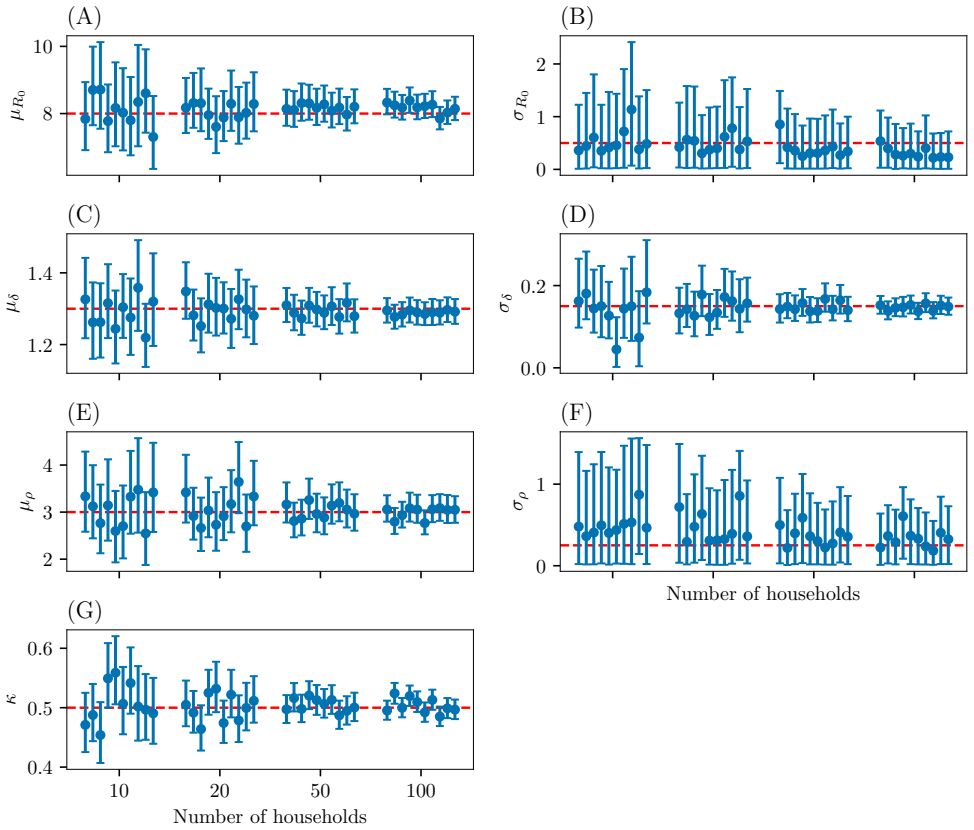
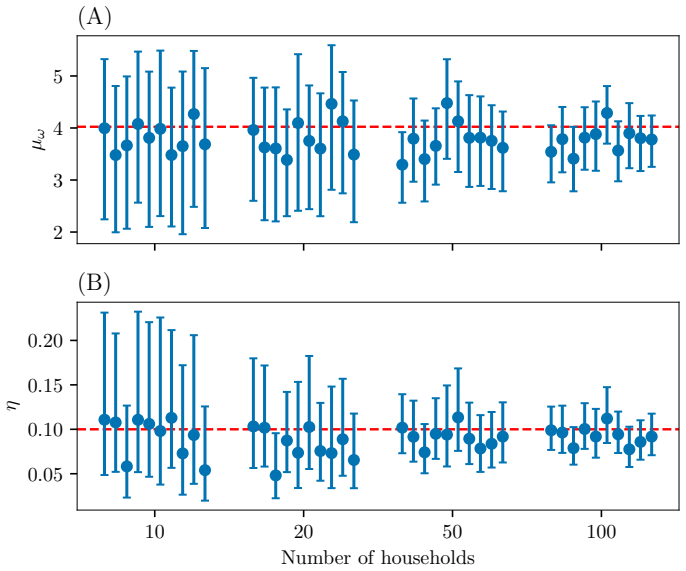




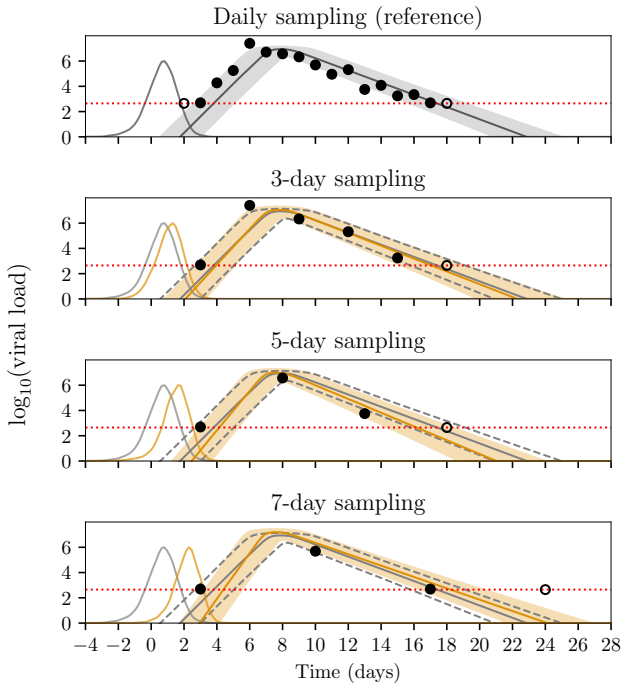

read the original abstract
Infectious disease dynamics operate across multiple biological scales, with within-host viral dynamics being a key driver of between-host transmission. However, while models that explicitly link these scales exist, none have been developed with statistical inference as a primary goal. In this paper we propose a multiscale model that jointly captures heterogeneous individual-level viral load trajectories and stochastic household transmission, and develop efficient inference methods to fit it to data. Since full joint inference is computationally difficult, we employ a cut approach that passes information from the within-host to the between-host model but not vice versa. This enables the data on viral loads to inform the transmission parameters such as the infection times and symptom onset thresholds. We evaluate the framework on simulated household outbreak data, assessing parameter recovery, computational efficiency, and the effect of viral load sampling frequency on inference quality. Parameter recovery is unbiased when the sampling frequency of the viral loads is high enough. When sampling is sparse, some bias is introduced, but incorporating external viral load data can mitigate this.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript proposes a multiscale Bayesian model integrating heterogeneous individual-level viral load trajectories with stochastic household transmission, using a one-way cut inference procedure to pass information from the within-host model to the between-host model without feedback. This enables estimation of transmission parameters such as infection times and symptom onset thresholds. The approach is evaluated solely on simulated household outbreak data, with claims of unbiased parameter recovery at high viral-load sampling frequencies and bias mitigation via external data when sampling is sparse.
Significance. If the cut approximation is shown to be robust beyond the simulated setting, the work offers a practical route to linking within-host and between-host scales for infectious disease modeling, with direct implications for improving estimates of key epidemiological quantities like infection timing.
major comments (2)
- [Inference Methods] The central claim of unbiased recovery of infection times and symptom-onset thresholds rests on the cut procedure (described in the inference section) that feeds point summaries or marginal posteriors from the within-host model into the household transmission model without feedback. The manuscript supplies no analytic bound on the resulting approximation error nor a small-scale comparison to full joint inference, which is load-bearing for the unbiasedness result when the two scales may be more tightly coupled than assumed.
- [Simulation Study] All parameter-recovery results (including the high-sampling-frequency unbiasedness statement) are obtained from data generated from the identical model used for fitting. This design cannot reveal distortion under realistic misspecification or when within-host and between-host dynamics are not cleanly separable, undermining the general claim that the cut produces accurate transmission-parameter estimates.
minor comments (2)
- [Abstract] The abstract states that 'parameter recovery is unbiased when the sampling frequency of the viral loads is high enough' but provides no quantitative threshold for 'high enough' nor any error-bar or coverage details from the simulations.
- [Results] Computational-efficiency claims are mentioned but lack concrete metrics (e.g., wall-clock time, effective sample size) or comparisons to alternative samplers.
Simulated Author's Rebuttal
We thank the referee for their constructive comments. We address each major point below, acknowledging the limitations of the cut approximation and the simulation design while clarifying the scope of our claims. We will make targeted revisions to improve transparency without altering the core methodology or results.
read point-by-point responses
-
Referee: [Inference Methods] The central claim of unbiased recovery of infection times and symptom-onset thresholds rests on the cut procedure (described in the inference section) that feeds point summaries or marginal posteriors from the within-host model into the household transmission model without feedback. The manuscript supplies no analytic bound on the resulting approximation error nor a small-scale comparison to full joint inference, which is load-bearing for the unbiasedness result when the two scales may be more tightly coupled than assumed.
Authors: We agree that the cut procedure is an approximation whose error lacks an analytic bound in the manuscript, and that a direct comparison to full joint inference would be valuable for assessing performance when scales are tightly coupled. Full joint inference is computationally prohibitive for the full model, which motivated the cut approach as described in the inference section. Our simulations demonstrate unbiased recovery of transmission parameters under frequent sampling when the model is correctly specified. We will revise the manuscript to include an expanded discussion of the cut approximation, its assumptions, and potential limitations, and we will add a small-scale numerical comparison on a reduced model where joint inference is feasible. revision: partial
-
Referee: [Simulation Study] All parameter-recovery results (including the high-sampling-frequency unbiasedness statement) are obtained from data generated from the identical model used for fitting. This design cannot reveal distortion under realistic misspecification or when within-host and between-host dynamics are not cleanly separable, undermining the general claim that the cut produces accurate transmission-parameter estimates.
Authors: We acknowledge that the simulation study generates data from the same model used for fitting, which is standard practice for validating inference procedures but does not address misspecification or non-separable dynamics. The manuscript claims unbiased recovery specifically when viral-load sampling is frequent and the model assumptions hold; it does not assert general robustness to misspecification. We will revise the text to explicitly qualify the unbiasedness result as holding under correct model specification, add a limitations paragraph discussing potential biases under misspecification, and note that robustness checks are left for future work. revision: partial
- Deriving a general analytic bound on the approximation error induced by the cut procedure for this multiscale model.
- Conducting a full-scale comparison between cut and joint inference due to prohibitive computational cost.
Circularity Check
No significant circularity detected in the multiscale inference derivation.
full rationale
The paper defines a joint multiscale generative model for viral loads and household transmission, then applies a standard one-directional cut approximation to enable tractable inference by passing within-host summaries to the transmission model without feedback. Parameter recovery is validated on data simulated from the same model, which is a conventional external check rather than a self-referential fit. No equations reduce a claimed prediction to a fitted input by construction, no uniqueness theorems are imported from self-citations, and no ansatz or renaming of known results is presented as a derivation. The central claims rest on the explicit modeling choices and simulation benchmarks, which remain independent of the target posterior.
Axiom & Free-Parameter Ledger
free parameters (2)
- infection times and symptom onset thresholds
- transmission rate parameters
axioms (2)
- domain assumption Viral load trajectories can be modeled as heterogeneous individual-level processes that drive transmission probability.
- ad hoc to paper One-way cut inference preserves unbiased estimation of transmission quantities.
Reference graph
Works this paper leans on
-
[1]
Infectious Disease Modelling 2, 128–142
A primer on stochastic epidemic models: Formulation, numerical simulation, and analysis. Infectious Disease Modelling 2, 128–142. doi:10.1016/j.idm.2017.03.001. Almocera, A.E.S., Nguyen, V.K., Hernandez-Vargas, E.A.,
-
[2]
Journal of Mathematical Biology 77, 1035–1057
Multiscale model within-host and between- host for viral infectious diseases. Journal of Mathematical Biology 77, 1035–1057. doi:10.1007/s00285 -018-1241-y. Amos, B.,
-
[3]
Foundations and Trends in Machine Learning 16, 592–732
Tutorial on Amortized Optimization. Foundations and Trends in Machine Learning 16, 592–732. doi:10.1561/2200000102. Ashcroft, P., Lehtinen, S., Bonhoeffer, S.,
-
[4]
Test-trace-isolate-quarantine (TTIQ) intervention strategies after symptomatic COVID-19 case identification. PLOS ONE 17, e0263597. doi:10.1371/journal.pone .0263597. Baccam, P., Beauchemin, C., Macken, C.A., Hayden, F.G., Perelson, A.S.,
-
[5]
Journal of Virology 80, 7590–7599
Kinetics of Influenza A Virus Infection in Humans. Journal of Virology 80, 7590–7599. doi:10.1128/JVI.01623-05. Barbour, A.D., Hamza, K., Kaspi, H., Klebaner, F.C.,
-
[6]
Advances in Applied Probability 47, 1190–1211
EscapefromtheboundaryinMarkovpopulation processes. Advances in Applied Probability 47, 1190–1211. doi:10.1239/aap/1449859806. Bédard, M.,
-
[7]
Stochastic Processes and their Applications 118, 2198–2222
Optimal acceptance rates for Metropolis algorithms: Moving beyond 0.234. Stochastic Processes and their Applications 118, 2198–2222. doi:10.1016/j.spa.2007.12.005. Bezanson, J., Edelman, A., Karpinski, S., Shah, V.B.,
-
[8]
Julia: A Fresh Approach to Numerical Com- puting. SIAM Review 59, 65–98. doi:10.1137/141000671. 31 Black, A.J., Geard, N., McCaw, J.M., McVernon, J., Ross, J.V.,
-
[9]
Epidemics 19, 61–73
Characterising pandemic severity and transmissibility from data collected during first few hundred studies. Epidemics 19, 61–73. doi:10.1 016/j.epidem.2017.01.004. Boddington, N.L., Charlett, A., Elgohari, S., Byers, C., Coughlan, L., Vilaplana, T.G., Whillock, R., Sinnathamby, M., Panagiotopoulos, N., Letley, L., MacDonald, P., Vivancos, R., Edeghere, O....
2017
-
[10]
Bulletin of the World Health Organization 99, 178–189
Epidemiological and clinical characteristics of early COVID-19 cases, United Kingdom of Great Britain and Northern Ireland. Bulletin of the World Health Organization 99, 178–189. doi:10.2471/BL T.20.265603. Bradbury, J., Frostig, R., Hawkins, P., Johnson, M.J., Leary, C., Maclaurin, D., Necula, G., Paszke, A., Van- derPlas, J., Wanderman-Milne, S., Zhang, Q.,
-
[11]
doi:10.1186/s12916-021-02220-0. Cleary, B., Hay, J.A., Blumenstiel, B., Harden, M., Cipicchio, M., Bezney, J., Simonton, B., Hong, D., Senghore, M., Sesay, A.K., Gabriel, S., Regev, A., Mina, M.J.,
-
[12]
Science Translational Medicine 13, eabf1568
Using viral load and epidemic dynamics to optimize pooled testing in resource-constrained settings. Science Translational Medicine 13, eabf1568. doi:10.1126/scitranslmed.abf1568. Cormen, T.H., Leiserson, C.E., Rivest, R.L., Stein, C.,
-
[13]
Mathematical methods for scaling from within-host to population-scale in infectious disease systems. Epidemics 45, 100724. doi:10.1016/j.epid em.2023.100724. Garira, W.,
-
[14]
Infectious Disease Modelling 3, 176–191
A primer on multiscale modelling of infectious disease systems. Infectious Disease Modelling 3, 176–191. doi:10.1016/j.idm.2018.09.005. Gelman, A., Carlin, J.B., Stern, H.S., Dunson, D.B., Vehtari, A., Rubin, D.B.,
-
[15]
Weak convergence and optimal scaling of random walk Metropolis algorithms. The Annals of Applied Probability 7, 110–120. doi:10.1214/aoap/1034625254. Goyal, A., Reeves, D.B., Schiffer, J.T.,
-
[16]
JournalofTheRoyalSocietyInterface19, 20210811
Multi-scale modelling reveals that early super-spreader events arealikelycontributortonovelvariantpredominance. JournalofTheRoyalSocietyInterface19, 20210811. doi:10.1098/rsif.2021.0811. Handel, A., Rohani, P.,
-
[17]
Philosophical Transactions of the Royal Society B: Biological Sciences 370, 20140302
Crossing the scale from within-host infection dynamics to between-host transmission fitness: A discussion of current assumptions and knowledge. Philosophical Transactions of the Royal Society B: Biological Sciences 370, 20140302. doi:10.1098/rstb.2014.0302. Hay, J.A., Kennedy-Shaffer, L., Kanjilal, S., Lennon, N.J., Gabriel, S.B., Lipsitch, M., Mina, M.J.,
-
[18]
Temporal dynamics in viral shedding and transmissibility of COVID-19. Nature Medicine 26, 672–675. doi:10.1038/s41591-020-0869-5. 32 House, T., Riley, H., Pellis, L., Pouwels, K.B., Bacon, S., Eidukas, A., Jahanshahi, K., Eggo, R.M., Sarah Walker, A.,
-
[19]
Statistical Methods in Medical Research 31, 1738–1756
Inferring risks of coronavirus transmission from community household data. Statistical Methods in Medical Research 31, 1738–1756. doi:10.1177/09622802211055853. .id (informed decisions),
-
[20]
Proceedings of the National Academy of Sciences 118, e2111477118
In vivo kinetics of SARS-CoV-2 infection and its relationship with a person’s infectiousness. Proceedings of the National Academy of Sciences 118, e2111477118. doi:10.1073/pnas.2111477118. Keeling, M.J., Rohani, P.,
-
[21]
Princeton University Press, Princeton, NJ
Modeling Infectious Diseases in Humans and Animals. Princeton University Press, Princeton, NJ. Kissler, S.M., Fauver, J.R., Mack, C., Olesen, S.W., Tai, C., Shiue, K.Y., Kalinich, C.C., Jednak, S., Ott, I.M., Vogels, C.B.F., Wohlgemuth, J., Weisberger, J., DiFiori, J., Anderson, D.J., Mancell, J., Ho, D.D., Grubaugh, N.D., Grad, Y.H., 2021a. Viral dynamic...
-
[22]
Viral Dynamics of SARS-CoV-2 Variants in Vaccinated and Unvaccinated Persons
Kissler, S.M., Fauver, J.R., Mack, C., Tai, C.G., Breban, M.I., Watkins, A.E., Samant, R.M., Anderson, D.J., Metti, J., Khullar, G., Baits, R., MacKay, M., Daisy, S., Baker, T., Dudley, J.T., Mason, C.E., Ho, D.D., Grubaugh, N.D., Grad, Y.H., 2021b. Viral Dynamics of SARS-CoV-2 Variants in Vaccinated and Unvaccinated Persons. New England Journal of Medici...
-
[23]
Estimating epidemic exponential growth rate and basic reproduction number. Infectious Disease Modelling 5, 129–141. doi:10.1016/j.idm.2019.12.009. Madewell, Z.J., Yang, Y., Longini, I.M., Halloran, M.E., Dean, N.E.,
-
[24]
Household Secondary Attack Rates of SARS-CoV-2 by Variant and Vaccination Status. JAMA Network Open 5, e229317. doi:10.100 1/jamanetworkopen.2022.9317. Marc, A., Kerioui, M., Blanquart, F., Bertrand, J., Mitjà, O., Corbacho-Monné, M., Marks, M., Guedj, J.,
-
[25]
Quantifying the relationship between SARS-CoV-2 viral load and infectiousness. eLife 10, e69302. doi:10.7554/eLife.69302. Marcato, A.J., Black, A.J., Walker, C.R., Morris, D., Meagher, N., Price, D.J., McVernon, J., 2022a. Learnings from the Australian first few X household transmission project for COVID-19. The Lancet Regional Health - Western Pacific
-
[26]
Marcato, A.J., Fielding, J.E., Crooks, K., Massey, P.D., Le, L.V., Bergeri, I., McVernon, J., 2022b
doi:10.1016/j.lanwpc.2022.100573. Marcato, A.J., Fielding, J.E., Crooks, K., Massey, P.D., Le, L.V., Bergeri, I., McVernon, J., 2022b. The on- going value of first few X studies for COVID-19 in the Western Pacific Region. Western Pacific Surveillance and Response Journal : WPSAR 13, 1–3. doi:10.5365/wpsar.2022.13.1.873. Margiotta, R.G., Sozio, E., Del Ben...
-
[27]
33 Marino, J., Cvitkovic, M., Yue, Y.,
doi:10.3389/fimmu.2024.1452638. 33 Marino, J., Cvitkovic, M., Yue, Y.,
-
[28]
Modelling: Understanding pandemics and how to control them. Epidemics 39, 100588. doi:10.1016/j.epidem.2022.100588. Martinez-Corral, R., Nam, K.M., DePace, A.H., Gunawardena, J.,
-
[29]
Proceedings of the National Academy of Sciences 121, e2318329121
The Hill function is the universal Hopfield barrier for sharpness of input–output responses. Proceedings of the National Academy of Sciences 121, e2318329121. doi:10.1073/pnas.2318329121. McLean, E., Pebody, R.G., Campbell, C., Chamberland, M., Hawkins, C., Nguyen-Van-Tam, J.S., Oliver, I., Smith, G.E., Ihekweazu, C., Bracebridge, S., Maguire, H., Harris,...
-
[30]
Epidemiology and Infection 138, 1531–1541
Pandemic (H1N1) 2009 influenza in the UK: Clinical and epidemiological findings from the first few hundred (FF100) cases. Epidemiology and Infection 138, 1531–1541. doi:10.1017/S0950268810001366. Morris, D., Maclean, J., Black, A.J.,
-
[31]
Morris, D.J., Kennedy, L., Black, A.J.,
doi:10.1007/s00285-024-02132-6. Morris, D.J., Kennedy, L., Black, A.J.,
-
[32]
PLOS Computational Biology 21, e1013775
Randomtime-shiftapproximationenableshierarchicalBayesian inference of mechanistic within-host viral dynamics models on large datasets. PLOS Computational Biology 21, e1013775. doi:10.1371/journal.pcbi.1013775. Pasupathy, R.,
-
[33]
Wiley Encyclopedia of Operations Research and Management Science doi:10.1002/9780470400531.eorms0356
Generating Nonhomogeneous Poisson Processes. Wiley Encyclopedia of Operations Research and Management Science doi:10.1002/9780470400531.eorms0356. Perelson, A.S.,
-
[34]
Nature Reviews Immunology 2, 28–36
Modelling viral and immune system dynamics. Nature Reviews Immunology 2, 28–36. doi:10.1038/nri700. Puhach, O., Meyer, B., Eckerle, I.,
-
[35]
Nature Reviews Microbiology 21, 147–161
SARS-CoV-2 viral load and shedding kinetics. Nature Reviews Microbiology 21, 147–161. doi:10.1038/s41579-022-00822-w. Reynolds, D.,
-
[36]
Gaussian Mixture Models, in: Encyclopedia of Biometrics. Springer, Boston, MA, pp. 659–663. doi:10.1007/978-0-387-73003-5_196. Schultze, J.L., Aschenbrenner, A.C.,
-
[37]
COVID-19 and the human innate immune system. Cell 184, 1671–1692. doi:10.1016/j.cell.2021.02.029. Serena, L., Marzolla, M., D’Angelo, G., Ferretti, S.,
-
[38]
Simulation Modelling Practice and Theory 127, 102780
A review of multilevel modeling and simulation for human mobility and behavior. Simulation Modelling Practice and Theory 127, 102780. doi:10.1016/ j.simpat.2023.102780. Smith, A.P., Moquin, D.J., Bernhauerova, V., Smith, A.M.,
-
[39]
Journal of Theoretical Biology 602–603, 112061
Efficient coupling of within-and between-host infectious disease dynamics. Journal of Theoretical Biology 602–603, 112061. doi:10.1016/j.jtbi.2025.112061. Stan Development Team,
-
[40]
A note on generation times in epidemic models. Mathematical Biosciences 208, 300–311. doi:10.1016/j.mbs.2006.10.010. 34 Swallow, B., Birrell, P., Blake, J., Burgman, M., Challenor, P., Coffeng, L.E., Dawid, P., De Angelis, D., Goldstein, M., Hemming, V., Marion, G., McKinley, T.J., Overton, C.E., Panovska-Griffiths, J., Pellis, L., Probert, W., Shea, K., ...
-
[41]
Challenges in estimation, uncertainty quantification and elicitation for pandemic modelling. Epidemics 38, 100547. doi:10.1016/j.epidem.2022.100547. Tatsukawa, Y., Arefin, M.R., Kuga, K., Tanimoto, J.,
-
[42]
Rank-Normalization, Folding, and Localization: An Improved R^ for Assessing Convergence of MCMC (with Discussion). Bayesian Analysis 16, 667–718. doi:10.1214/20-BA1221. Wang, X., Wang, S., Wang, J., Rong, L.,
-
[43]
doi:10.1007/s11538-022-01058-8. World Health Organization,
-
[44]
Yin, Y., Flegg, J.A., Flegg, M.B.,
The first few X cases and contacts (FFX) investigation protocol for coronavirus disease 2019 (COVID-19), version 2.2.https://www.who.int/publications/i/item/th e-first-few-x-cases-and-contacts-(-ffx)-investigation-protocol-for-coronavirus-disease -2019-(-covid-19)-version-2.2. Yin, Y., Flegg, J.A., Flegg, M.B.,
2019
-
[45]
Journal of Theoretical Biology 612, 112194
Accurate stochastic simulation algorithm for multiscale models of infectious diseases. Journal of Theoretical Biology 612, 112194. doi:10.1016/j.jtbi.2025.112194. Zitzmann, C., Ke, R., Ribeiro, R.M., Perelson, A.S.,
-
[46]
doi:10.1371/journal.pc bi.1011437
How robust are estimates of key parameters in standard viral dynamic models? PLOS Computational Biology 20, e1011437. doi:10.1371/journal.pc bi.1011437. 35
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.