Recognition: unknown
Conditional Monte Carlo Tree Diffusion for Designing Cell-Type-Specific and Biologically Faithful Regulatory DNA
Pith reviewed 2026-05-09 22:52 UTC · model grok-4.3
The pith
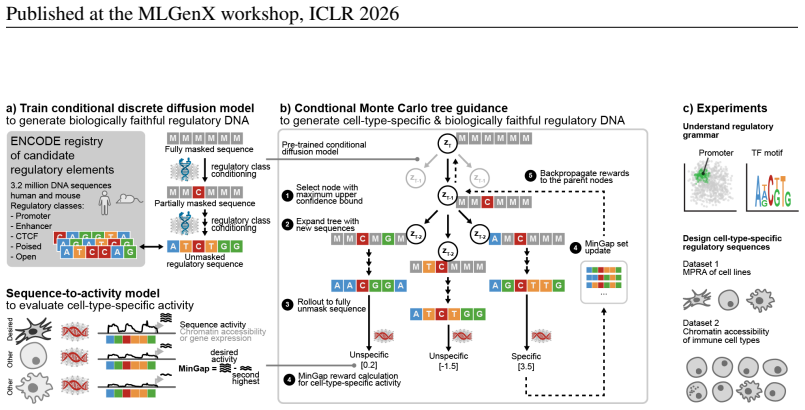
DNA-CRAFT combines class-conditioned discrete diffusion with Monte Carlo tree guidance to generate regulatory DNA sequences that achieve high cell-type specificity while preserving natural biological grammar.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
DNA-CRAFT first trains a class-conditioned discrete diffusion model on millions of natural regulatory sequences to capture cell-class-specific grammars, then uses conditional Monte Carlo tree guidance during inference to maximize differential regulatory activity between desired and undesired cell types, producing sequences with high predicted specificity and fidelity to genome patterns.
What carries the argument
Conditional Monte Carlo tree guidance, an inference-time algorithm that steers the sampling of a class-conditioned discrete diffusion model to optimize the difference in predicted regulatory activity between target and non-target cell types.
If this is right
- Produces regulatory sequences for enhancers and promoters that better respect natural grammar while increasing target-cell activity.
- Delivers improved specificity against undesired cell types compared to diffusion-only, autoregressive, or optimization baselines.
- Supports design tasks across human cell lines and immune cell types with measurable gains in the specificity-fidelity trade-off.
- Offers a scalable inference procedure that can be applied after training the base diffusion model on large genomic registries.
Where Pith is reading between the lines
- If experimental validation confirms the predictions, the method could shorten the design cycle for synthetic promoters used in targeted gene delivery.
- The tree-guidance step may transfer to other conditional sequence-generation problems where both activity and sequence realism must be controlled.
- Additional constraints such as chromatin state or evolutionary conservation could be folded into the same guidance procedure to further refine outputs.
Load-bearing premise
The model's learned regulatory grammars from ENCODE data remain valid under tree guidance and its activity predictions correspond to actual cellular function without introducing non-natural artifacts.
What would settle it
Lab assays that measure actual transcriptional output of the generated sequences in both target and off-target human cell types to test whether the predicted specificity and fidelity appear in living cells.
Figures




read the original abstract
Designing regulatory DNA elements with precise cell-type-specific activity is broadly relevant for cell engineering and gene therapy. Deep generative models can generate functional gene-regulatory elements, but existing methods struggle to achieve high specificity against undesired cell types while adhering to the genome's natural regulatory grammar. Here, we introduce DNA-CRAFT, a generative framework that integrates class-conditioned discrete diffusion with Monte Carlo tree search to design cell-type-specific and biologically faithful regulatory elements. We first train a discrete diffusion model on the ENCODE registry of 3.2 million candidate regulatory elements. Second, we condition the model to learn class-specific regulatory grammars of naturally occurring DNA sequences, including enhancers and promoters. Third, we employ conditional Monte Carlo tree guidance, an inference-time alignment algorithm designed to maximize the differential regulatory activity between desired and undesired cell types. By benchmarking DNA-CRAFT on regulatory sequence design tasks for human cell lines and immune cell types, we demonstrate that our model generates sequences with high predicted cell-type-specific activity and biological fidelity, achieving the best trade-offs compared to methods that use diffusion, autoregressive models, and gradient-based optimization.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript introduces DNA-CRAFT, a generative framework that trains a class-conditioned discrete diffusion model on the ENCODE registry of 3.2 million regulatory elements to learn cell-type-specific grammars for enhancers and promoters, then applies conditional Monte Carlo tree search guidance at inference time to maximize differential regulatory activity between desired and undesired cell types. Benchmarking on human cell lines and immune cell types is claimed to yield sequences with high predicted cell-type-specific activity and biological fidelity, outperforming diffusion, autoregressive, and gradient-based baselines.
Significance. If the performance and fidelity claims are substantiated, the integration of diffusion models with inference-time MCTS guidance could offer a practical advance for designing synthetic regulatory elements in gene therapy and cell engineering, addressing the specificity limitations of prior generative approaches while aiming to preserve natural sequence constraints.
major comments (2)
- Abstract: the central claim of 'best trade-offs' and 'high predicted cell-type-specific activity and biological fidelity' is asserted without any quantitative metrics, baseline details, evaluation protocols, statistical tests, or effect sizes. This absence prevents verification that the data support superiority over the compared methods.
- Methods/Results (guidance and evaluation sections): the conditional Monte Carlo tree guidance optimizes against a differential activity predictor, yet the manuscript provides no description of an independent held-out activity oracle, cross-validation on model-generated sequences, or wet-lab assays. This leaves open the risk that optimized sequences exploit predictor artifacts rather than reflecting genuine regulatory grammar, which is load-bearing for the 'biologically faithful' claim.
Simulated Author's Rebuttal
We thank the referee for the constructive comments. We address each major point below and will revise the manuscript to improve clarity and transparency.
read point-by-point responses
-
Referee: Abstract: the central claim of 'best trade-offs' and 'high predicted cell-type-specific activity and biological fidelity' is asserted without any quantitative metrics, baseline details, evaluation protocols, statistical tests, or effect sizes. This absence prevents verification that the data support superiority over the compared methods.
Authors: We agree that the abstract would be strengthened by including quantitative support. In the revision we will add specific metrics (e.g., mean differential activity scores and fidelity measures versus baselines), a brief statement of the evaluation protocol, and reference to statistical comparisons. revision: yes
-
Referee: Methods/Results (guidance and evaluation sections): the conditional Monte Carlo tree guidance optimizes against a differential activity predictor, yet the manuscript provides no description of an independent held-out activity oracle, cross-validation on model-generated sequences, or wet-lab assays. This leaves open the risk that optimized sequences exploit predictor artifacts rather than reflecting genuine regulatory grammar, which is load-bearing for the 'biologically faithful' claim.
Authors: The study is computational and relies on predictors trained on ENCODE data. We will expand the methods section to detail the cross-validation performed on the activity predictors and to state explicitly that no independent held-out oracle or wet-lab validation was conducted. We will also add a limitations paragraph acknowledging the possibility of predictor artifacts while noting that training on natural sequence distributions and the use of fidelity metrics (sequence-level similarity to known regulatory elements) provide partial safeguards. The 'biologically faithful' language will be qualified accordingly. revision: partial
Circularity Check
No significant circularity; training on external ENCODE data with inference-time guidance
full rationale
The paper's core chain trains a class-conditioned discrete diffusion model on the external ENCODE registry of 3.2 million elements, then applies conditional Monte Carlo tree search at inference time to maximize a differential activity score. No quoted equations, self-citations, or steps in the abstract or described framework reduce the claimed cell-type-specific activity or biological fidelity to a fitted parameter defined by the result itself, a self-referential definition, or a load-bearing self-citation. Benchmarking against diffusion, autoregressive, and gradient baselines is presented as an empirical comparison on held-out tasks, without evidence that the performance metric collapses to the training inputs by construction. This satisfies the default expectation of a self-contained derivation.
Axiom & Free-Parameter Ledger
axioms (2)
- domain assumption The ENCODE registry of 3.2 million candidate regulatory elements is representative of natural class-specific regulatory grammars.
- ad hoc to paper Conditional Monte Carlo tree guidance can be applied at inference time to maximize differential regulatory activity between cell types.
Reference graph
Works this paper leans on
-
[1]
URL https://openreview.net/forum?fileGuid=3xgr169o12oUrbxS& id=HklxbgBKvr&ref=https%3A%2F%2Fgithubhelp.com. Jacob Austin, Daniel D. Johnson, Jonathan Ho, Daniel Tarlow, and Rianne van den Berg. Structured Denoising Diffusion Models in Discrete State-Spaces, February 2023. URL http://arxiv. org/abs/2107.03006. arXiv:2107.03006 [cs]. Pavel Avdeyev, Chenlai ...
-
[2]
URL https://www.nature.com/articles/ s41588-025-02441-6
doi: 10.1038/s41588-025-02441-6. URL https://www.nature.com/articles/ s41588-025-02441-6. Publisher: Nature Publishing Group. 10 Published at the MLGenX workshop, ICLR 2026 Bernardo P. de Almeida, Christoph Schaub, Michaela Pagani, Stefano Secchia, Eileen E. M. Furlong, and Alexander Stark. Targeted design of synthetic enhancers for selected tis- sues in ...
-
[3]
URL https://www.nature.com/articles/ s41586-023-06905-9
doi: 10.1038/s41586-023-06905-9. URL https://www.nature.com/articles/ s41586-023-06905-9. Publisher: Nature Publishing Group. Bieke Decaesteker, Geertrui Denecker, Christophe Van Neste, Emmy M. Dolman, Wouter Van Loocke, Moritz Gartlgruber, Carolina Nunes, Fanny De Vloed, Pauline Depuydt, Karen Verboom, Dries Rombaut, Siebe Loontiens, Jolien De Wyn, Walee...
-
[4]
URL https://www.nature.com/articles/ s12276-024-01233-y
doi: 10.1038/s12276-024-01233-y. URL https://www.nature.com/articles/ s12276-024-01233-y. Publisher: Nature Publishing Group. Sager J Gosai, Rodrigo I Castro, Natalia Fuentes, John C Butts, Kousuke Mouri, Michael Alasoadura, Susan Kales, Thanh Thanh L Nguyen, Ramil R Noche, Arya S Rao, et al. Machine-guided design of cell-type-targeting cis-regulatory ele...
-
[5]
URLhttps://doi.org/10.1093/nar/gkaf205
doi: 10.1093/nar/gkaf205. URLhttps://doi.org/10.1093/nar/gkaf205. Moksh Jain, Emmanuel Bengio, Alex Hernandez-Garcia, Jarrid Rector-Brooks, Bonaventure FP Dossou, Chanakya Ajit Ekbote, Jie Fu, Tianyu Zhang, Michael Kilgour, Dinghuai Zhang, et al. Biological sequence design with gflownets. InInternational Conference on Machine Learning, pp. 9786–9801. PMLR...
-
[6]
URLhttp://arxiv.org/abs/2408.08252. arXiv:2408.08252 [cs]. Yang Eric Li, Sebastian Preissl, Michael Miller, Nicholas D. Johnson, Zihan Wang, Henry Jiao, Chenxu Zhu, Zhaoning Wang, Yang Xie, Olivier Poirion, Colin Kern, Antonio Pinto-Duarte, Wei Tian, Kimberly Siletti, Nora Emerson, Julia Osteen, Jacinta Lucero, Lin Lin, Qian Yang, Quan Zhu, Nathan Zemke, ...
-
[7]
doi: 10.1016/j.molcel.2017.08.026
ISSN 1097-2765. doi: 10.1016/j.molcel.2017.08.026. URL https://www.cell.com/ molecular-cell/abstract/S1097-2765(17)30624-X. Publisher: Elsevier. Subham Sekhar Sahoo, Marianne Arriola, Yair Schiff, Aaron Gokaslan, Edgar Marroquin, Justin T. Chiu, Alexander Rush, and V olodymyr Kuleshov. Simple and Effective Masked Diffu- sion Language Models, November 2024...
-
[8]
URL https://www.nature.com/articles/ s41467-021-24932-w
doi: 10.1038/s41467-021-24932-w. URL https://www.nature.com/articles/ s41467-021-24932-w. Publisher: Nature Publishing Group. Chenyu Wang, Masatoshi Uehara, Yichun He, Amy Wang, Tommaso Biancalani, Avantika Lal, Tommi Jaakkola, Sergey Levine, Hanchen Wang, and Aviv Regev. Fine-Tuning Discrete Diffusion Models via Reward Optimization with Applications to D...
-
[9]
doi: 10.1523/JNEUROSCI.1077-11.2011
ISSN 0270-6474, 1529-2401. doi: 10.1523/JNEUROSCI.1077-11.2011. URL https: //www.jneurosci.org/content/31/35/12413. Publisher: Society for Neuroscience Section: Articles. Jinshou Yang, Feihan Zhou, Xiyuan Luo, Yuan Fang, Xing Wang, Xiaohong Liu, Ruiling Xiao, Decheng Jiang, Yuemeng Tang, Gang Yang, Lei You, and Yupei Zhao. Enhancer reprogramming: critical...
-
[10]
doi: 10.1038/s41420-025-02366-3
ISSN 2058-7716. doi: 10.1038/s41420-025-02366-3. URL https://www.nature. com/articles/s41420-025-02366-3. Publisher: Nature Publishing Group. Zhao Yang, Bing Su, Chuan Cao, and Ji-Rong Wen. Regulatory dna sequence design with reinforce- ment learning. InThe Thirteenth International Conference on Learning Representations. Hongyong Zhang, Zechen Li, Yanmei ...
-
[11]
Homology Edges:An edge connects a human cCRE and a mouse cCRE if they share a sequence alignment of>100bp, determined via the hg38-mm10 syntenic nets Kent et al. (2002)
2002
-
[12]
19 Published at the MLGenX workshop, ICLR 2026 We computed the connected components ofG to define independent sequence clusters
Overlap Edges:Within a single species, an edge connects any two cCREs that share sequence similarity of more than100bp. 19 Published at the MLGenX workshop, ICLR 2026 We computed the connected components ofG to define independent sequence clusters. These clusters were randomly partitioned into training (90%), validation (5%), and test (5%) sets, ensuring ...
2026
-
[13]
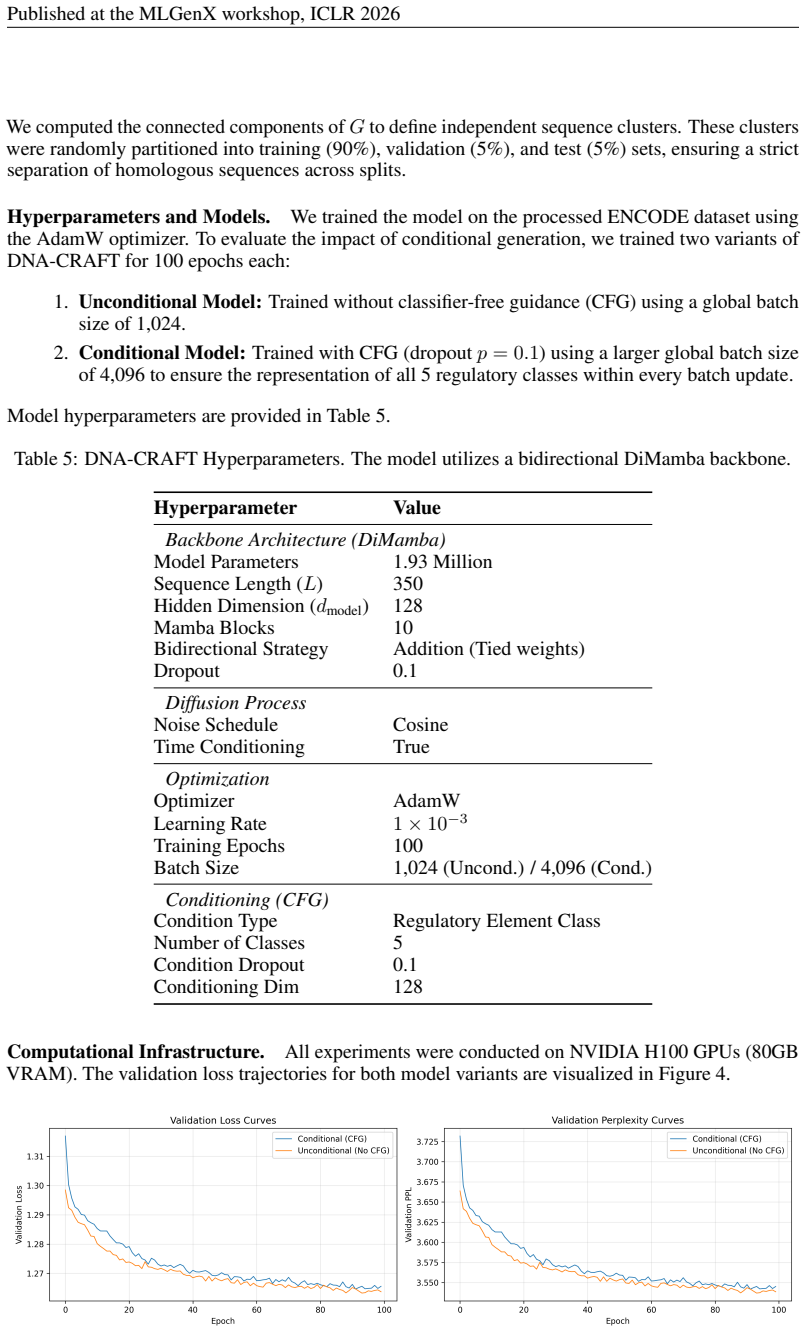
Unconditional Model:Trained without classifier-free guidance (CFG) using a global batch size of 1,024
-
[14]
Model hyperparameters are provided in Table 5
Conditional Model:Trained with CFG (dropout p= 0.1 ) using a larger global batch size of 4,096 to ensure the representation of all 5 regulatory classes within every batch update. Model hyperparameters are provided in Table 5. Table 5: DNA-CRAFT Hyperparameters. The model utilizes a bidirectional DiMamba backbone. Hyperparameter Value Backbone Architecture...
2026
-
[15]
We implemented a custom differentiable wrapper to compute MinGap scores fromDesign-Model’s output and backpropagate gradients directly to the input sequence representation
Ledidi:We initialized the optimization with 128 random DNA sequences. We implemented a custom differentiable wrapper to compute MinGap scores fromDesign-Model’s output and backpropagate gradients directly to the input sequence representation. Optimization was conducted independently for each sequence for a maximum of 20,000 steps to ensure convergence. 21...
2026
-
[16]
We performed 128 parallel diffusion inference steps using our pre-trained unconditional diffusion model
Classifier Guidance (CG):We used DNA-CRAFT’s unconditional base diffusion model, trained on the entire ENCODE registry of regulatory elements without further fine-tuning for any experiments. We performed 128 parallel diffusion inference steps using our pre-trained unconditional diffusion model. Gradients were computed via the Design-Model wrapper to guide...
2026
-
[17]
The sampling process tracked 128 particles and applied a resampling parameter ofα= 0.5
Sequential Monte Carlo (SMC):We used the same unconditional base diffusion model and MinGap wrapper as the CG method. The sampling process tracked 128 particles and applied a resampling parameter ofα= 0.5
-
[18]
We applied a guidance scale of γ= 1000 and a resampling parameter of α= 0.5
Twisted Diffusion Sampling (TDS):We followed the same setup as CG and SMC for this baseline. We applied a guidance scale of γ= 1000 and a resampling parameter of α= 0.5
-
[19]
We generated 128 sequences per cell type with conditional sampling (γ= 4.0)
D3 (Discrete Denoising Diffusion):We trained the discrete diffusion model utilizing the transformer backbone (2M parameters) for a 100 epochs on the MPRA dataset (∼700,000 sequences) with the cell type activity as classes. We generated 128 sequences per cell type with conditional sampling (γ= 4.0)
-
[20]
We fine-tuned three separate models (one for each cell line: HepG2, K562, SK-N-SH) for 100 optimization steps
Ctrl-DNA:We followed the original protocol and hyperparameters specified for each target cell type. We fine-tuned three separate models (one for each cell line: HepG2, K562, SK-N-SH) for 100 optimization steps. The top 128 sequences from each fine-tuning run were selected for evaluation. We note that Ctrl-DNA’s base autoregressive model Nguyen et al. (202...
2023
-
[21]
For the HepG2 cell line, we utilized the provided pre-trained checkpoint
DRAKES:We followed the protocol described in the original publication. For the HepG2 cell line, we utilized the provided pre-trained checkpoint. Similarly, we fine-tuned two separate models for K562 and SK-N-SH. We generated sequences using the respective fine-tuned models. We note that DRAKES fine-tunes its diffusion model using reward gradients computed...
-
[22]
Candidate sequences were generated using conditional Monte Carlo tree guidance
DNA-CRAFT (Ours):We employ DNA-CRAFT’s class-conditioned base diffusion model, which is trained on the ENCODE dataset without further fine-tuning for all experiments. Candidate sequences were generated using conditional Monte Carlo tree guidance. We selected the final candidate from the MinGap set G∗ at the end of the tree search. Table 8 details the spec...
2025
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.