Recognition: unknown
Enabling Biomolecular Simulations with Neural Network Potentials in GROMACS
Pith reviewed 2026-05-08 13:10 UTC · model grok-4.3
The pith
An interface in GROMACS lets PyTorch-trained neural network potentials supply energies and forces during molecular dynamics runs.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
The interface supplies a flexible definition of model inputs and outputs that lets any PyTorch neural network potential deliver energies and forces to GROMACS during an MD step. This integration occurs for chosen subsystems or the entire system and preserves access to GROMACS advanced sampling and free-energy methods, so the neural potentials can be used in production biomolecular calculations without leaving the standard simulation environment.
What carries the argument
The NNP interface, which defines a general set of model inputs and outputs so that PyTorch neural-network inference can be called inside GROMACS force evaluation and integrator loops.
If this is right
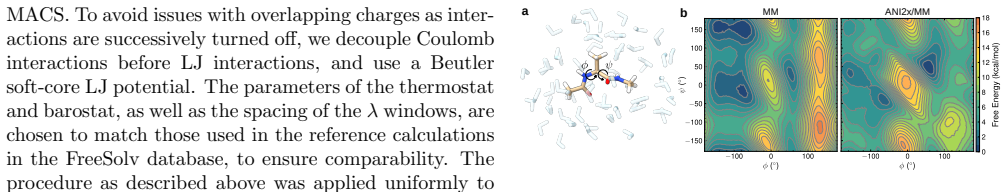
- Enhanced sampling of peptide torsional landscapes can be performed directly with neural potentials inside GROMACS.
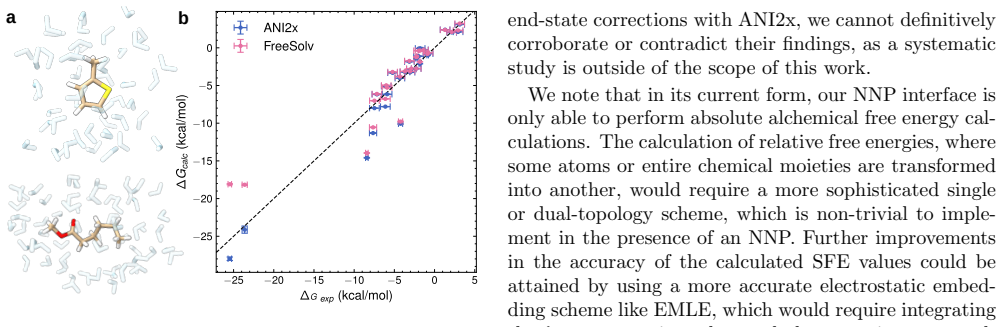
- Absolute solvation free energies can be computed by treating solute and solvent regions with different potentials.
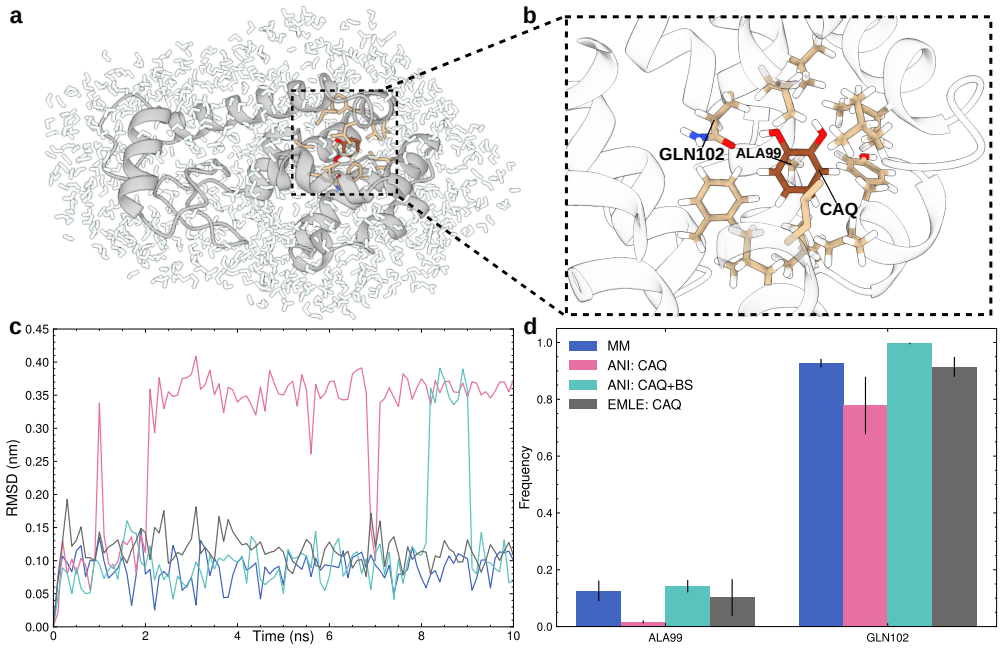
- Protein-ligand binding simulations can use neural potentials on the ligand or binding site while keeping the rest of the system under standard force fields.
- Performance benchmarks on water boxes give concrete timing data for several neural network architectures inside the same code base.
Where Pith is reading between the lines
- The same interface pattern could be adapted to other machine-learning frameworks beyond PyTorch, widening the set of available potentials.
- Because GROMACS already supports large-scale parallel runs, the interface makes neural potentials immediately usable for systems with tens or hundreds of thousands of atoms.
- Free-energy calculations that combine neural potentials with alchemical transformations become feasible without custom code for each new potential.
Load-bearing premise
Neural network potentials must return energies and forces that remain numerically stable and physically consistent when mixed with GROMACS force calculations and time-stepping routines.
What would settle it
Run a constant-energy simulation with the interface active and observe whether total energy drifts or the trajectory becomes unstable within a few thousand steps.
Figures


read the original abstract
Neural network potentials (NNPs) are rapidly changing the landscape of state-of-the-art molecular dynamics (MD) simulations. To make full use of this development, the community needs flexible, easy-to-use interfaces firmly integrated with existing methodologies. To address this, we here present an interface for hybrid machine learning/molecular mechanics (ML/MM) simulations implemented in the widely used MD code GROMACS. The interface enables NNPs trained in the PyTorch framework to contribute energies and forces during MD simulations, either for selected subsets or entire molecular systems. By defining a flexible set of model inputs and outputs, the interface is agnostic to specific NNP architectures and can accommodate a wide range of descriptor-based and message-passing models. In particular, the design integrates NNP inference seamlessly into the extensive GROMACS molecular simulation ecosystem, providing users with the capability to straightforwardly combine NNPs with existing advanced sampling and free energy workflows. We demonstrate the capabilities of the interface using several representative applications, including enhanced sampling of peptide torsional free energy landscapes, absolute solvation free energy calculations, and protein--ligand simulations. We also run performance benchmarks on water boxes for several different NNP architectures. Our interface is available in recent GROMACS releases, and we believe it will provide a practical foundation for incorporating machine learning potentials into production MD simulations of biomolecular systems.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The paper presents an interface implemented in GROMACS that enables the use of neural network potentials (NNPs) trained in the PyTorch framework for providing energies and forces in molecular dynamics simulations. The interface supports both hybrid ML/MM setups for selected subsets and full NNP simulations for entire systems. It is designed to be agnostic to specific NNP architectures and integrates with GROMACS's advanced sampling and free energy workflows. The capabilities are demonstrated through applications such as enhanced sampling of peptide torsional free energy landscapes, absolute solvation free energy calculations, protein-ligand simulations, and performance benchmarks on water boxes.
Significance. If the interface performs as described, this work has high significance for the field of computational physics and biomolecular simulation. It provides a practical means to incorporate state-of-the-art machine learning potentials into a popular MD engine, facilitating their use in production-level simulations involving complex systems and advanced techniques. The availability in recent GROMACS releases and the architecture-agnostic design are particular strengths that could accelerate adoption of NNPs.
minor comments (2)
- [Abstract] The abstract could briefly note any observed computational overhead or limitations from the benchmarks to give readers a more complete picture of practical use.
- [Performance benchmarks] Performance benchmarks would be strengthened by including a direct comparison to standard classical force fields on the same water-box systems to contextualize the NNP overhead.
Simulated Author's Rebuttal
We thank the referee for their positive evaluation of the manuscript, recognition of its significance, and recommendation to accept. No major comments were raised.
Circularity Check
No significant circularity; self-contained engineering implementation
full rationale
The manuscript describes the design and implementation of a GROMACS interface for hybrid ML/MM simulations using external PyTorch-trained neural network potentials. It provides explicit code-level details on model input/output handling, integration with existing GROMACS workflows for sampling and free-energy calculations, and validates the interface through independent demonstrations (peptide torsional sampling, solvation free energies, protein-ligand runs) plus architecture-specific performance benchmarks on water boxes. No load-bearing step derives a result from its own fitted parameters, self-citations, or ansatz; the claims rest on the engineering artifacts and external model compatibility rather than any internal derivation chain.
Axiom & Free-Parameter Ledger
axioms (1)
- domain assumption Standard assumptions of classical molecular dynamics (Newtonian mechanics, force fields, integrators)
Reference graph
Works this paper leans on
-
[1]
& Levitt, M
Warshel, A. & Levitt, M. Theoretical Studies of En- zymic Reactions: Dielectric, Electrostatic and Steric Stabilization of the Carbonium Ion in the Reaction of Lysozyme.Journal of Molecular Biology103, 227–249 (May 1976)
1976
-
[2]
& Truhlar, D
Lin, H. & Truhlar, D. G. QM/MM: What Have We Learned, Where Are We, and Where Do We Go from Here?Theoretical Chemistry Accounts117, 185–199 (Feb. 2007)
2007
-
[3]
Senn, H. M. & Thiel, W. QM/MM Methods for Biomolecular Systems.Angewandte Chemie Inter- national Edition48,1198–1229 (2009)
2009
-
[4]
T.et al.Machine Learning Force Fields
Unke, O. T.et al.Machine Learning Force Fields. Chemical Reviews121,10142–10186 (Aug. 2021)
2021
-
[5]
Four Generations of High-Dimensional Neural Network Potentials.Chemical Reviews121, 10037–10072 (Aug
Behler, J. Four Generations of High-Dimensional Neural Network Potentials.Chemical Reviews121, 10037–10072 (Aug. 2021)
2021
-
[6]
& Parrinello, M
Behler, J. & Parrinello, M. Generalized Neural- Network Representation of High-Dimensional Potential-Energy Surfaces.Physical Review Letters 98,146401 (Apr. 2007)
2007
-
[7]
S., Isayev, O
Smith, J. S., Isayev, O. & Roitberg, A. E. ANI-1: An Extensible Neural Network Potential with DFT Ac- curacy at Force Field Computational Cost.Chemi- cal Science8,3192–3203 (Mar. 2017)
2017
-
[8]
Devereux, C.et al.Extending the Applicability of the ANI Deep Learning Molecular Potential to Sul- fur and Halogens.Journal of Chemical Theory and Computation16,4192–4202 (July 2020)
2020
-
[9]
S., Leszczynski, J
Zubatyuk, R., Smith, J. S., Leszczynski, J. & Isayev, O. Accurate and Transferable Multitask Prediction of Chemical Properties with an Atoms-in-Molecules Neural Network.Science Advances5,eaav6490 (Aug. 2019)
2019
-
[10]
M., Zubatyuk, R
Anstine, D. M., Zubatyuk, R. & Isayev, O. AIM- Net2: A Neural Network Potential to Meet Your Neutral, Charged, Organic, and Elemental-Organic Needs.Chemical Science16,10228–10244 (June 2025)
2025
-
[11]
Batatia, I., Kov´ acs, D. P., Simm, G. N. C., Ortner, C. & Cs´ anyi, G.MACE: Higher Order Equivariant Message Passing Neural Networks for Fast and Ac- curate Force FieldsJan. 2023. arXiv:2206.07697 [stat]. 9
-
[12]
Unke, O. T. & Meuwly, M. PhysNet: A Neural Network for Predicting Energies, Forces, Dipole Moments, and Partial Charges.Journal of Chem- ical Theory and Computation15,3678–3693 (June 2019)
2019
-
[13]
Batzner, S.et al.E(3)-Equivariant Graph Neu- ral Networks for Data-Efficient and Accurate In- teratomic Potentials.Nature Communications13, 2453 (May 2022)
2022
-
[14]
T.et al.Biomolecular Dynamics with Machine-Learned Quantum-Mechanical Force Fields Trained on Diverse Chemical Fragments.Sci- ence Advances10,eadn4397 (Apr
Unke, O. T.et al.Biomolecular Dynamics with Machine-Learned Quantum-Mechanical Force Fields Trained on Diverse Chemical Fragments.Sci- ence Advances10,eadn4397 (Apr. 2024)
2024
-
[15]
P.et al.MACE-OFF: Short-Range Transferable Machine Learning Force Fields for Or- ganic Molecules.Journal of the American Chemical Society147,17598–17611 (May 2025)
Kov´ acs, D. P.et al.MACE-OFF: Short-Range Transferable Machine Learning Force Fields for Or- ganic Molecules.Journal of the American Chemical Society147,17598–17611 (May 2025)
2025
-
[16]
Kabylda, A.et al.Molecular Simulations with a Pretrained Neural Network and Universal Pairwise Force Fields.Journal of the American Chemical So- ciety147,33723–33734 (Sept. 2025)
2025
-
[17]
Machine-Learning Interatomic Poten- tials for Materials Science.Acta Materialia214, 116980 (Aug
Mishin, Y. Machine-Learning Interatomic Poten- tials for Materials Science.Acta Materialia214, 116980 (Aug. 2021)
2021
-
[18]
Musaelian, A.et al.Learning Local Equivariant Representations for Large-Scale Atomistic Dynam- ics.Nature Communications14,579 (Feb. 2023)
2023
-
[19]
& Zavadlav, J
Fuchs, P., Chen, W., Thaler, S. & Zavadlav, J. Chemtrain-Deploy: A Parallel and Scalable Frame- work for Machine Learning Potentials in Million- Atom MD Simulations.Journal of Chemical Theory and Computation21,7550–7560 (Aug. 2025)
2025
-
[20]
T.et al.SpookyNet: Learning Force Fields with Electronic Degrees of Freedom and Nonlo- cal Effects.Nature Communications12,7273 (Dec
Unke, O. T.et al.SpookyNet: Learning Force Fields with Electronic Degrees of Freedom and Nonlo- cal Effects.Nature Communications12,7273 (Dec. 2021)
2021
-
[21]
E., Toth, D
Yao, K., Herr, J. E., Toth, D. W., Mckintyre, R. & Parkhill, J. The TensorMol-0.1 Model Chem- istry: A Neural Network Augmented with Long- Range Physics.Chemical Science9,2261–2269 (Feb. 2018)
2018
-
[22]
W., Finkler, J
Ko, T. W., Finkler, J. A., Goedecker, S. & Behler, J. A Fourth-Generation High-Dimensional Neural Network Potential with Accurate Electrostatics In- cluding Non-Local Charge Transfer.Nature Com- munications12,398 (Jan. 2021)
2021
-
[23]
& de Gironcoli, S
Shaidu, Y., Pellegrini, F., K¨ u¸ c¨ ukbenli, E., Lot, R. & de Gironcoli, S. Incorporating Long-Range Electro- statics in Neural Network Potentials via Variational Charge Equilibration from Shortsighted Ingredi- ents.npj Computational Materials10,47 (Mar. 2024)
2024
-
[24]
Lahey, S.-L. J. & Rowley, C. N. Simulating Protein– Ligand Binding with Neural Network Potentials. Chemical Science11,2362–2368 (Mar. 2020)
2020
-
[25]
Rufa, D. A.et al. Towards Chemical Accuracy for Alchemical Free Energy Calculations with Hybrid Physics-Based Machine Learning / Molecular Me- chanics PotentialsJuly 2020. bioRxiv:2020 . 07 . 29.227959
2020
-
[26]
Saban´ es Zariquiey, F.et al.Enhancing Protein– Ligand Binding Affinity Predictions Using Neural Network Potentials.Journal of Chemical Informa- tion and Modeling64,1481–1485 (Mar. 2024)
2024
-
[27]
Karwounopoulos, J.et al.Insights and Challenges in Correcting Force Field Based Solvation Free En- ergies Using a Neural Network Potential.The Jour- nal of Physical Chemistry B128,6693–6703 (July 2024)
2024
-
[28]
Sch¨ utt, K
Gastegger, M., T. Sch¨ utt, K. & M¨ uller, K.-R. Machine Learning of Solvent Effects on Molecu- lar Spectra and Reactions.Chemical Science12, 11473–11483 (2021)
2021
-
[29]
A.et al.Advancing Multiscale Molecu- lar Modeling with Machine Learning-Derived Elec- trostatics.Journal of Chemical Theory and Com- putation(Mar
Semelak, J. A.et al.Advancing Multiscale Molecu- lar Modeling with Machine Learning-Derived Elec- trostatics.Journal of Chemical Theory and Com- putation(Mar. 2025)
2025
-
[30]
& Curutchet, C
Zinovjev, K. & Curutchet, C. Improved Description of Environment and Vibronic Effects with Electro- statically Embedded ML Potentials.The Journal of Physical Chemistry Letters16,774–781 (Jan. 2025)
2025
-
[31]
The Journal of Physical Chemistry B128,109–116 (Jan
Eastman, P.et al.OpenMM 8: Molecular Dynam- ics Simulation with Machine Learning Potentials. The Journal of Physical Chemistry B128,109–116 (Jan. 2024)
2024
-
[32]
Galvelis, R.et al.NNP/MM: Accelerating Molec- ular Dynamics Simulations with Machine Learn- ing Potentials and Molecular Mechanics.Journal of Chemical Information and Modeling63,5701–5708 (Sept. 2023)
2023
-
[33]
Electrostatic Embedding of Machine Learning Potentials.Journal of Chemical Theory and Computation19,1888–1897 (Mar
Zinovjev, K. Electrostatic Embedding of Machine Learning Potentials.Journal of Chemical Theory and Computation19,1888–1897 (Mar. 2023)
2023
-
[34]
Zinovjev, K.et al.Emle-Engine: A Flexible Elec- trostatic Machine Learning Embedding Package for Multiscale Molecular Dynamics Simulations.Jour- nal of Chemical Theory and Computation20,4514– 4522 (June 2024)
2024
-
[35]
& Riniker, S
Pultar, F., Th¨ urlemann, M., Gordiy, I., Doloszeski, E. & Riniker, S. Neural Network Potential with Multiresolution Approach Enables Accurate Pre- diction of Reaction Free Energies in Solution.Jour- nal of the American Chemical Society147,6835– 6856 (Feb. 2025). 10
2025
-
[36]
Paszke, A.et al.inProceedings of the 33rd Interna- tional Conference on Neural Information Process- ing Systems721, 8026–8037 (Dec. 2019)
2019
-
[37]
JAX: Composable Transforma- tions of Python+NumPy Programs2018
Bradbury, J.et al. JAX: Composable Transforma- tions of Python+NumPy Programs2018
-
[38]
Hjorth Larsen, A.et al.The Atomic Simulation Environment—a Python Library for Working with Atoms.Journal of Physics: Condensed Matter29, 273002 (June 2017)
2017
-
[39]
P.et al.LAMMPS - a Flexible Sim- ulation Tool for Particle-Based Materials Modeling at the Atomic, Meso, and Continuum Scales.Com- puter Physics Communications271,108171 (Feb
Thompson, A. P.et al.LAMMPS - a Flexible Sim- ulation Tool for Particle-Based Materials Modeling at the Atomic, Meso, and Continuum Scales.Com- puter Physics Communications271,108171 (Feb. 2022)
2022
-
[40]
A., Xue, J
Pickering, I., Semelak, J. A., Xue, J. & Roitberg, A. E. TorchANI-Amber: Bridging Neural Network Potentials and Classical Biomolecular Simulations. The Journal of Physical Chemistry B129,11927– 11938 (Nov. 2025)
2025
-
[41]
Zeng, J.et al.DeePMD-kit v3: A Multiple-Backend Framework for Machine Learning Potentials.Jour- nal of Chemical Theory and Computation21,4375– 4385 (May 2025)
2025
-
[42]
J.et al.GROMACS: High Perfor- mance Molecular Simulations through Multi-Level Parallelism from Laptops to Supercomputers.Soft- wareX1–2,19–25 (Sept
Abraham, M. J.et al.GROMACS: High Perfor- mance Molecular Simulations through Multi-Level Parallelism from Laptops to Supercomputers.Soft- wareX1–2,19–25 (Sept. 2015)
2015
-
[43]
Mobley, D. L. & Guthrie, J. P. FreeSolv: A Database of Experimental and Calculated Hydra- tion Free Energies, with Input Files.Journal of Computer-Aided Molecular Design28,711–720 (July 2014)
2014
-
[44]
Chmiela, S.et al.Machine Learning of Accurate Energy-Conserving Molecular Force Fields.Science Advances3,e1603015 (May 2017)
2017
-
[45]
S.et al.Assessment of Embedding Schemes in a Hybrid Machine Learning/Classical Potentials (ML/MM) Approach.Journal of Chem- ical Information and Modeling64,4047–4058 (May 2024)
Grassano, J. S.et al.Assessment of Embedding Schemes in a Hybrid Machine Learning/Classical Potentials (ML/MM) Approach.Journal of Chem- ical Information and Modeling64,4047–4058 (May 2024)
2024
-
[46]
& Slipchenko, L
Haghiri, S., Viquez Rojas, C., Bhat, S., Isayev, O. & Slipchenko, L. ANI/EFP: Modeling Long-Range Interactions in ANI Neural Network with Effective Fragment Potentials.Journal of Chemical Theory and Computation20,9138–9147 (Oct. 2024)
2024
-
[47]
& Oostenbrink, C
Lier, B., Poliak, P., Marquetand, P., Westermayr, J. & Oostenbrink, C. BuRNN: Buffer Region Neural Network Approach for Polarizable-Embedding Neu- ral Network/Molecular Mechanics Simulations.The Journal of Physical Chemistry Letters13,3812– 3818 (May 2022)
2022
-
[48]
Singh, U. C. & Kollman, P. A. A Combined Ab Ini- tio Quantum Mechanical and Molecular Mechanical Method for Carrying out Simulations on Complex Molecular Systems: Applications to the CH3Cl + Cl- Exchange Reaction and Gas Phase Protonation of Polyethers.Journal of Computational Chemistry 7,718–730 (1986)
1986
-
[49]
J., Albe, M., Bret, C., Proust-De Martin, F
Field, M. J., Albe, M., Bret, C., Proust-De Martin, F. & Thomas, A. The Dynamo Library for Molec- ular Simulations Using Hybrid Quantum Mechan- ical and Molecular Mechanical Potentials.Journal of Computational Chemistry21,1088–1100 (2000)
2000
-
[50]
& Golovin, A
Zlobin, A., Belyaeva, J. & Golovin, A. Challenges in Protein QM/MM Simulations with Intra-Backbone Link Atoms.Journal of Chemical Information and Modeling63,546–560 (Jan. 2023). 51.Reference Manual - GROMACS 2026.0 Docu- mentationhttps : / / manual . gromacs . org / documentation / current / reference - manual / index.html
2023
-
[51]
Gao, X., Ramezanghorbani, F., Isayev, O., Smith, J. S. & Roitberg, A. E. TorchANI: A Free and Open Source PyTorch-based Deep Learning Implementa- tion of the ANI Neural Network Potentials.Journal of Chemical Information and Modeling60,3408– 3415 (2020)
2020
-
[52]
& Roitberg, A
Pickering, I., Xue, J., Huddleston, K., Terrel, N. & Roitberg, A. E. TorchANI 2.0: An Extensible, High- Performance Library for the Design, Training, and Use of NN-IPs.Journal of Chemical Information and Modeling65,11656–11671 (Nov. 2025)
2025
-
[53]
P., Chodera, J
Eastman, P., Pritchard, B. P., Chodera, J. D. & Markland, T. E. Nutmeg and SPICE: Models and Data for Biomolecular Machine Learning.Journal of Chemical Theory and Computation20,8583– 8593 (Oct. 2024)
2024
-
[54]
& Hess, B
Lindahl, V., Lidmar, J. & Hess, B. Accelerated Weight Histogram Method for Exploring Free En- ergy Landscapes.The Journal of Chemical Physics 141,044110 (July 2014)
2014
-
[55]
M., Caldwell, J
Wang, J., Wolf, R. M., Caldwell, J. W., Kollman, P. A. & Case, D. A. Development and Testing of a General Amber Force Field.Journal of Computa- tional Chemistry25,1157–1174 (2004)
2004
-
[56]
L., Chandrasekhar, J., Madura, J
Jorgensen, W. L., Chandrasekhar, J., Madura, J. D., Impey, R. W. & Klein, M. L. Comparison of Simple Potential Functions for Simulating Liq- uid Water.The Journal of Chemical Physics79, 926–935 (July 1983)
1983
-
[57]
& Parrinello, M
Bussi, G., Donadio, D. & Parrinello, M. Canonical Sampling through Velocity Rescaling.The Journal of Chemical Physics126,014101 (Jan. 2007). 11
2007
-
[58]
& Bussi, G
Bernetti, M. & Bussi, G. Pressure Control Using Stochastic Cell Rescaling.The Journal of Chemical Physics153,114107 (Sept. 2020)
2020
-
[59]
Bennett, C. H. Efficient Estimation of Free En- ergy Differences from Monte Carlo Data.Journal of Computational Physics22,245–268 (Oct. 1976)
1976
-
[60]
Vanommeslaeghe, K.et al.CHARMM General Force Field: A Force Field for Drug-like Molecules Compatible with the CHARMM All-Atom Additive Biological Force Fields.Journal of Computational Chemistry31,671–690 (2010)
2010
-
[61]
Best, R. B.et al.Optimization of the Additive CHARMM All-Atom Protein Force Field Target- ing Improved Sampling of the Backboneϕ,ψand Side-Chainχ1 andχ2 Dihedral Angles.Journal of Chemical Theory and Computation8,3257–3273 (Sept. 2012)
2012
-
[62]
& Fiorucci, S
Bouysset, C. & Fiorucci, S. ProLIF: A Library to Encode Molecular Interactions as Fingerprints. Journal of Cheminformatics13,72 (Sept. 2021)
2021
-
[63]
& Kopple, K
Madison, V. & Kopple, K. D. Solvent-Dependent Conformational Distributions of Some Dipeptides. Journal of the American Chemical Society102, 4855–4863 (July 1980)
1980
-
[64]
Tobias, D. J. & Brooks, C. L. I. Conformational Equilibrium in the Alanine Dipeptide in the Gas Phase and Aqueous Solution: A Comparison of The- oretical Results.The Journal of Physical Chemistry 96,3864–3870 (Apr. 1992)
1992
-
[65]
Tian, C.et al.ff19SB: Amino-Acid-Specific Pro- tein Backbone Parameters Trained against Quan- tum Mechanics Energy Surfaces in Solution.Jour- nal of Chemical Theory and Computation16,528– 552 (Jan. 2020)
2020
-
[66]
Lundborg, M.et al.Skin Permeability Prediction with MD Simulation Sampling Spatial and Al- chemical Reaction Coordinates.Biophysical Jour- nal121,3837–3849 (Oct. 2022)
2022
-
[67]
Harry Moore, J., Cole, D. J. & Cs´ anyi, G. Com- puting Solvation Free Energies of Small Molecules with Experimental Accuracy.Journal of the Amer- ican Chemical Society(Jan. 2026)
2026
-
[68]
E.et al.Predicting Ligand Binding Affin- ity with Alchemical Free Energy Methods in a Polar Model Binding Site.Journal of Molecular Biology 394,747–763 (Dec
Boyce, S. E.et al.Predicting Ligand Binding Affin- ity with Alchemical Free Energy Methods in a Polar Model Binding Site.Journal of Molecular Biology 394,747–763 (Dec. 2009)
2009
-
[69]
J., Zhang, J., Klinman, J
Kulik, H. J., Zhang, J., Klinman, J. P. & Mart´ ınez, T. J. How Large Should the QM Region Be in QM/MM Calculations? The Case of Catechol O- Methyltransferase.The Journal of Physical Chem- istry B120,11381–11394 (Nov. 2016)
2016
-
[70]
V., Robey, R., Li, Y
Yokelson, D., Tkachenko, N. V., Robey, R., Li, Y. W. & Dub, P. A. Performance Analysis of CP2K Code for Ab Initio Molecular Dynamics on CPUs and GPUs.Journal of Chemical Information and Modeling62,2378–2386 (May 2022)
2022
-
[71]
& Riniker, S.AMP-BMS/MM: A Multiscale Neural Network Potential for the Fast and Accurate Simulation of Protein Dynamics and Enzymatic Re- actions2026
Th¨ urlemann, M., Pultar, F., Gordiy, I., Ruijsenaars, E. & Riniker, S.AMP-BMS/MM: A Multiscale Neural Network Potential for the Fast and Accurate Simulation of Protein Dynamics and Enzymatic Re- actions2026. ChemRxiv:10 . 26434 / chemrxiv - 2026-kx9w0. 12 Supporting Information S1: Solvation Free Energy Calculations on the FreeSolv database. ID ∆G exp σe...
2026
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.