Recognition: unknown
Supregraph: Enabling Information-Optimal Assembly Graph Representation of a Read Set
Pith reviewed 2026-05-08 12:51 UTC · model grok-4.3
The pith
Supregraphs provide a complete graph representation of error-free reads without information loss or artificial breaks.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
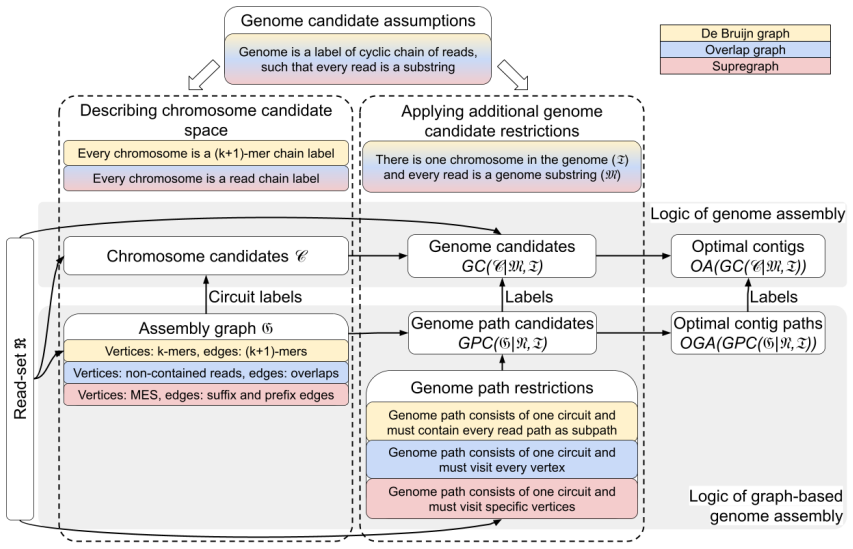
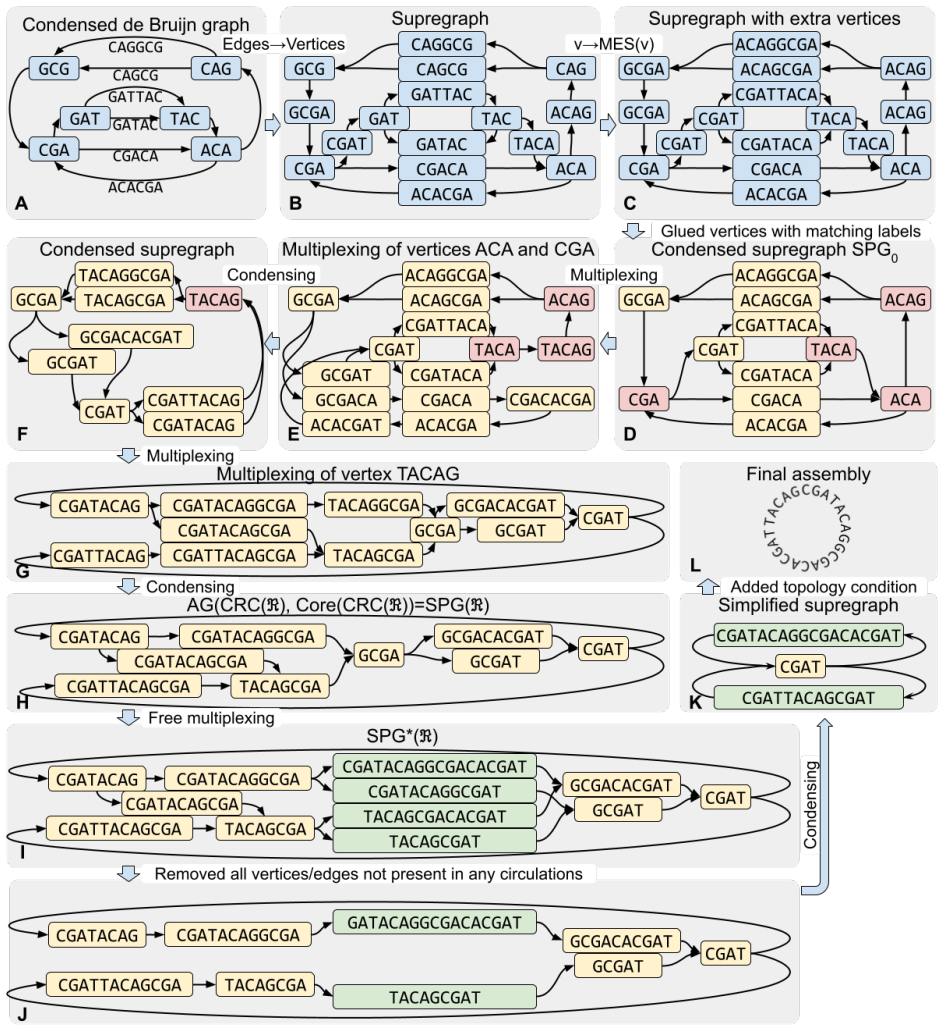
We prove that a correct representation of a read set exists in the form of a new class of assembly graphs, which we call supregraphs. Supregraphs can be constructed by iteratively transforming de Bruijn graphs using the multiplexing procedure. Under a set of natural assumptions, supregraphs provide a foundation for constructing theoretically optimal genome assemblies.
What carries the argument
Supregraphs, a class of assembly graphs constructed by multiplexing de Bruijn graphs to encode every overlap and every contained read without discarding data or introducing breaks.
If this is right
- Supregraphs preserve complete information from the read set where de Bruijn graphs lose details.
- They eliminate the artificial breaks that arise when overlap graphs discard contained reads.
- They are obtained from de Bruijn graphs by applying the multiplexing procedure used in existing assemblers.
- Under the natural assumptions they support the construction of theoretically optimal genome assemblies.
Where Pith is reading between the lines
- Assembly pipelines could be restructured to operate directly on supregraphs to reduce information loss at the graph-construction stage.
- The multiplexing transformation offers a concrete route for adapting current de Bruijn-based tools to produce information-complete graphs.
- Extending the error-free model to include realistic error rates would be a direct next step for practical use.
Load-bearing premise
The model assumes all reads are error-free, and the optimality result further depends on an unspecified collection of natural assumptions whose validity is not demonstrated.
What would settle it
Build a supregraph from an error-free read set generated from a known reference genome that contains contained reads, then check whether every possible consistent assembly path appears in the graph without missing overlaps or forced breaks.
Figures



read the original abstract
The first step in any genome assembly algorithm entails the conversion from the domain of strings and overlaps to the language of graphs and paths, typically using one of the two conventional methods: de Bruijn graphs or overlap graphs. However, both standard approaches are known to have limitations. De Bruijn graphs fail to represent complete information from reads, while the overlap graphs often produce artificial breaks in contigs due to the necessity to discard contained reads as a preliminary step. In this work we present a mathematical model for genome assembly that provides a formal framework to determine what constitutes a correct conversion of a read set into an assembly graph under the assumption of error-free reads. We prove that a correct representation of a read set exists in the form of a new class of assembly graphs, which we call supregraphs. We show that supregraphs can be constructed by iteratively transforming de Bruijn graphs using the multiplexing procedure, previously employed in the genome assemblers LJA and Verkko. Finally, we demonstrate that, under a set of natural assumptions, supregraphs provide a foundation for constructing theoretically optimal genome assemblies.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The paper introduces supregraphs as a new class of assembly graphs that correctly and completely represent error-free read sets. It provides a mathematical model for what constitutes a correct read-to-graph conversion, proves existence of such representations, demonstrates an explicit construction by iteratively applying multiplexing to de Bruijn graphs (building on LJA and Verkko), and asserts that under a set of natural assumptions these graphs enable theoretically optimal genome assemblies.
Significance. If the optimality claim can be made precise, the work would supply a useful theoretical foundation for genome assembly by eliminating information loss inherent in de Bruijn graphs and the artificial breaks caused by discarding contained reads in overlap graphs. The existence proof and the constructive multiplexing procedure are clear strengths that could guide future assembler development.
major comments (1)
- [Abstract and optimality section] Abstract and the optimality discussion (likely §5 or the final section): the headline claim that supregraphs 'provide a foundation for constructing theoretically optimal genome assemblies' is conditioned on 'a set of natural assumptions' that are neither enumerated nor shown to be sufficient. Because this conditional step is load-bearing for the paper's significance, the assumptions (e.g., restrictions on repeat structure, coverage uniformity, or path uniqueness) must be stated explicitly and their sufficiency for optimality demonstrated.
minor comments (1)
- [Abstract] The abstract would be clearer if it briefly indicated the scope of the 'natural assumptions' even at a high level.
Simulated Author's Rebuttal
We thank the referee for the constructive review and for recognizing the potential of supregraphs as a theoretical foundation. We address the single major comment below.
read point-by-point responses
-
Referee: [Abstract and optimality section] Abstract and the optimality discussion (likely §5 or the final section): the headline claim that supregraphs 'provide a foundation for constructing theoretically optimal genome assemblies' is conditioned on 'a set of natural assumptions' that are neither enumerated nor shown to be sufficient. Because this conditional step is load-bearing for the paper's significance, the assumptions (e.g., restrictions on repeat structure, coverage uniformity, or path uniqueness) must be stated explicitly and their sufficiency for optimality demonstrated.
Authors: We agree that the conditional optimality claim requires explicit enumeration and justification. In the revised manuscript we will insert a dedicated paragraph in the optimality discussion that lists the assumptions in full: (1) reads are error-free, (2) coverage is uniform and sufficient to resolve all unique paths in the multiplexed graph, and (3) repeat structures do not exceed the resolving power of the given read lengths (i.e., no unresolvable tangles remain after multiplexing). We will also add a concise argument showing sufficiency: under these conditions the existence proof and the iterative multiplexing construction together guarantee that every read corresponds to a unique path and that the genome is recovered without information loss or artificial breaks. This revision directly strengthens the load-bearing step identified by the referee. revision: yes
Circularity Check
No significant circularity; core proof and definition are independent of inputs
full rationale
The paper introduces a mathematical model for converting read sets to assembly graphs under error-free reads, defines supregraphs as a new class, and claims to prove that a correct representation exists in this form. It further shows construction via iterative transformation of de Bruijn graphs using the multiplexing procedure from prior assemblers LJA and Verkko. This reference is a standard method citation rather than a load-bearing justification that reduces the existence proof to self-citation or prior unverified results. No equations, fitted parameters, self-definitional constructs, or uniqueness theorems imported from overlapping authors appear in the derivation. The conditional optimality claim under unspecified 'natural assumptions' affects claim strength but does not create circularity by making any result equivalent to its inputs by construction. The derivation chain remains self-contained against external benchmarks.
Axiom & Free-Parameter Ledger
axioms (1)
- domain assumption Reads are error-free
invented entities (1)
-
supregraph
no independent evidence
Reference graph
Works this paper leans on
-
[1]
Accurate circular consensus long-read sequencing improves variant detection and assembly of a human genome
Aaron M Wenger, Paul Peluso, William J Rowell, Pi-Chuan Chang, Richard J Hall, Gregory T Con- cepcion, Jana Ebler, Arkarachai Fungtammasan, Alexey Kolesnikov, Nathan D Olson, et al. Accurate circular consensus long-read sequencing improves variant detection and assembly of a human genome. Nature Biotechnology, 37(10):1155–1162, 2019
2019
-
[2]
Pair consensus decoding improves accuracy of neural network basecallers for nanopore sequencing.Genome biology, 22(1):1–13, 2021
Jordi Silvestre-Ryan and Ian Holmes. Pair consensus decoding improves accuracy of neural network basecallers for nanopore sequencing.Genome biology, 22(1):1–13, 2021
2021
-
[3]
The complete sequence of a human genome.Science, 376(6588):44–53, 2022
Sergey Nurk, Sergey Koren, Arang Rhie, Mikko Rautiainen, Andrey V Bzikadze, Alla Mikheenko, Mitchell R Vollger, Nicolas Altemose, Lev Uralsky, Ariel Gershman, et al. The complete sequence of a human genome.Science, 376(6588):44–53, 2022
2022
-
[4]
A draft human pangenome reference.Nature, 617(7960):312–324, 2023
Wen-Wei Liao, Mobin Asri, Jana Ebler, Daniel Doerr, Marina Haukness, Glenn Hickey, Shuangjia Lu, Julian K Lucas, Jean Monlong, Haley J Abel, et al. A draft human pangenome reference.Nature, 617(7960):312–324, 2023
2023
-
[5]
Complete sequencing of ape genomes.Nature, 641:401–418, 2025
Dahyun Yoo, Arang Rhie, Prashanth Hebbar, et al. Complete sequencing of ape genomes.Nature, 641:401–418, 2025
2025
-
[6]
Closeread: a tool for assessing assembly errors in immunoglobulin loci applied to vertebrate long-read genome assemblies
Yilei Zhu, Camila Watson, Yana Safonova, Matthew Pennell, and Anton Bankevich. Closeread: a tool for assessing assembly errors in immunoglobulin loci applied to vertebrate long-read genome assemblies. Genome Biology, 26(1):131, 2025
2025
-
[7]
Combinatorial algorithms for dna sequence assembly.Algo- rithmica, 13(1):7–51, 1995
John D Kececioglu and Eugene W Myers. Combinatorial algorithms for dna sequence assembly.Algo- rithmica, 13(1):7–51, 1995
1995
-
[8]
Toward simplifying and accurately formulating fragment assembly.Journal of Com- putational Biology, 2(2):275–290, 1995
Eugene W Myers. Toward simplifying and accurately formulating fragment assembly.Journal of Com- putational Biology, 2(2):275–290, 1995
1995
-
[9]
Computability of models for se- quence assembly
Paul Medvedev, Katerina Georgiou, Gene Myers, and Michael Brudno. Computability of models for se- quence assembly. InInternational Workshop on Algorithms in Bioinformatics, pages 289–301. Springer, 2007
2007
-
[10]
Information-optimal genome assembly via sparse read-overlap graphs.Bioinformatics, 32(17):i494–i502, 2016
Ilan Shomorony, Seung H Kim, Thomas A Courtade, and David N Tse. Information-optimal genome assembly via sparse read-overlap graphs.Bioinformatics, 32(17):i494–i502, 2016
2016
-
[11]
Safe and complete contig assembly through omnitigs.Journal of Computational Biology, 24(6):590–602, 2017
Alexandru I Tomescu and Paul Medvedev. Safe and complete contig assembly through omnitigs.Journal of Computational Biology, 24(6):590–602, 2017
2017
-
[12]
Assembler artifacts include misassembly because of unsafe unitigs and underassembly because of bidirected graphs.Genome Research, 32(9):1746–1753, 2022
A Rahman and P Medvedev. Assembler artifacts include misassembly because of unsafe unitigs and underassembly because of bidirected graphs.Genome Research, 32(9):1746–1753, 2022
2022
-
[13]
An eulerian path approach to dna fragment assembly.Proceedings of the National Academy of Sciences, 98(17):9748–9753, 2001
Pavel A Pevzner, Haixu Tang, and Michael S Waterman. An eulerian path approach to dna fragment assembly.Proceedings of the National Academy of Sciences, 98(17):9748–9753, 2001
2001
-
[14]
The fragment assembly string graph.Bioinformatics, 21(suppl 2):ii79–ii85, 2005
Eugene W Myers. The fragment assembly string graph.Bioinformatics, 21(suppl 2):ii79–ii85, 2005. 18
2005
-
[15]
Exploiting sparse- ness in de novo genome assembly.BMC Bioinformatics, 13(6):1–11, 2012
Chengxi Ye, Zhanshan Sam Ma, Charles H Cannon, Mihai Pop, and Douglas W Yu. Exploiting sparse- ness in de novo genome assembly.BMC Bioinformatics, 13(6):1–11, 2012
2012
-
[16]
Assembly of long error-prone reads using repeat graphs.Nature Biotechnology, 37(5):540–546, 2019
Mikhail Kolmogorov, Jeffrey Yuan, Yu Lin, and Pavel A Pevzner. Assembly of long error-prone reads using repeat graphs.Nature Biotechnology, 37(5):540–546, 2019
2019
-
[17]
Genome assembly in the telomere-to-telomere era.Nature Reviews Genetics, 25(9):658–670, 2024
Heng Li and Richard Durbin. Genome assembly in the telomere-to-telomere era.Nature Reviews Genetics, 25(9):658–670, 2024
2024
-
[18]
Overlap-based genome assembly from variable-length reads
Jonathan Hui, Ilan Shomorony, Kannan Ramchandran, and Thomas A Courtade. Overlap-based genome assembly from variable-length reads. In2016 IEEE International Symposium on Information Theory (ISIT), pages 1018–1022. IEEE, 2016
2016
-
[19]
Coverage-preserving sparsification of overlap graphs for long-read assembly.Bioinformatics, 39(3):btad124, 2023
Chirag Jain. Coverage-preserving sparsification of overlap graphs for long-read assembly.Bioinformatics, 39(3):btad124, 2023
2023
-
[20]
Telomere-to-telomere assembly by preserving contained reads.Genome Research, 34(11):1908–1918, 2024
Siddharth S Kamath, Mridul Bindra, Debajyoti Pal, and Chirag Jain. Telomere-to-telomere assembly by preserving contained reads.Genome Research, 34(11):1908–1918, 2024
1908
-
[21]
Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm.Nature Methods, 18(2):170–175, 2021
Haoyu Cheng, Gregory T Concepcion, Xiaowen Feng, Haowen Zhang, and Heng Li. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm.Nature Methods, 18(2):170–175, 2021
2021
-
[22]
Multiplex de bruijn graphs enable genome assembly from long, high-fidelity reads.Nature Biotechnology, 40(7):1075–1081, 2022
Anton Bankevich, Andrey V Bzikadze, Mikhail Kolmogorov, Dmitry Antipov, and Pavel A Pevzner. Multiplex de bruijn graphs enable genome assembly from long, high-fidelity reads.Nature Biotechnology, 40(7):1075–1081, 2022
2022
-
[23]
Telomere-to-telomere assembly of diploid chro- mosomes with verkko.Nature Biotechnology, 41(10):1474–1482, 2023
Mikko Rautiainen, Sergey Nurk, Brian P Walenz, Glennis A Logsdon, David Porubsky, Arang Rhie, Evan E Eichler, Adam M Phillippy, and Sergey Koren. Telomere-to-telomere assembly of diploid chro- mosomes with verkko.Nature Biotechnology, 41(10):1474–1482, 2023
2023
-
[24]
Papert.Counter-Free Automata
Robert McNaughton and Seymour A. Papert.Counter-Free Automata. MIT Research Monograph. MIT Press, Cambridge, MA, 1971
1971
-
[25]
Families of locally testable languages
Kangho Kim, Robert McNaughton, and Brigitte McCloskey. Families of locally testable languages. Theoretical Computer Science, 234(1–2):47–75, 2000
2000
-
[26]
A characterization of strictly locally testable languages and its application to subsemigroups of a free semigroup.Information and Control, 44(3):300–319, 1980
Aldo De Luca and Antonio Restivo. A characterization of strictly locally testable languages and its application to subsemigroups of a free semigroup.Information and Control, 44(3):300–319, 1980
1980
-
[27]
Versatile and open software for comparing large genomes.Genome biology, 5(2):1–9, 2004
Stefan Kurtz, Adam Phillippy, Arthur L Delcher, Michael Smoot, Martin Shumway, Corina Antonescu, and Steven L Salzberg. Versatile and open software for comparing large genomes.Genome biology, 5(2):1–9, 2004
2004
-
[28]
Idba–a practical iterative de bruijn graph de novo assembler
Yu Peng, Henry CM Leung, and Francis YL Chin. Idba–a practical iterative de bruijn graph de novo assembler. InAnnual international conference on research in computational molecular biology, pages 426–440. Springer, 2010
2010
-
[29]
Gurevich, Mikhail Dvorkin, Alexander S
Anton Bankevich, Sergey Nurk, Dmitry Antipov, Alexey A. Gurevich, Mikhail Dvorkin, Alexander S. Kulikov, Valery M. Lesin, Sergey I. Nikolenko, Son Pham, Andrey D. Prjibelski, Alexey V. Pyshkin, Alexander V. Sirotkin, Nikolay Vyahhi, Glenn Tesler, Max A. Alekseyev, and Pavel A. Pevzner. Spades: A new genome assembly algorithm and its applications to single...
2012
-
[30]
genome tangle
Evgeny Kapun and Fedor Tsarev. De bruijn superwalk with multiplicities problem is np-hard.BMC Bioinformatics, 14(5):1–7, 2013. 19 Appendix Appendix A1. Bidirected strings and graphs While we traditionally represent DNA as a sequence of nucleotides, the real physical DNA molecule contains two chains with nucleotide sequences reverse-complementary to each o...
2013
-
[31]
For any two walksPandQinG,Label(P)is a prefix/suffix/substring/equal ofLabel(Q)iffPis a prefix walk/suffix walk/subwalk/equal ofQ
-
[32]
Proof: (1)IfLabel(P) is a proper infix ofLabel(Q), then by non-redundancy ofG,Pis a subwalk of Q
For any vertex or edgevand walkPinG, the number of occurrences ofvinPis the same as the number of occurrences ofLabel(v)inLabel(P). Proof: (1)IfLabel(P) is a proper infix ofLabel(Q), then by non-redundancy ofG,Pis a subwalk of Q. IfLabel(P) is a proper suffix ofLabel(Q), consider a walkQ ′ obtained fromQby appending one additional edgeeto its end (such an...
-
[33]
If a vertex inGhas an outgoing suffix/incoming prefix edge, it can have no other outgoing/incoming edges
-
[34]
If a vertexvinGis a proper prefix/suffix of vertexu, then there exists unique path fromvtou/from utovthat consists only of prefix/suffix edges
-
[35]
All vertices in a non-redundant graph have different labels
-
[36]
Vertex label can not be a proper infix of another vertex label. 23
-
[37]
Proof: (1)Assume that a vertexvhas both a suffix and a non-suffix outgoing edge, denoteds=vwand n=vu, respectively
For any walkPinGthere exists unique closed walkP c and unqiue open walkP o such thatLabel(P) = Label(Po) =Label(P c).P o can be obtained fromP c by removing all prefix/suffix edges from the start/end ofP c. Proof: (1)Assume that a vertexvhas both a suffix and a non-suffix outgoing edge, denoteds=vwand n=vu, respectively. The label of the path consisting s...
-
[38]
For any two closed walksPandQinG, such thatLabel(P)is a substring/prefix/suffix ofLabel(Q), Pis a subwalk/prefix walk/suffix walk/equal ofQ
-
[39]
For a closed walkP, any walkQ, such thatLabel(Q)is a substring ofLabel(P)must be a subwalk of P
-
[40]
For any two open walksPandQinG, wherePis non-trivial (contains at least one edge),Label(P) is a prefix/suffix/substring ofLabel(Q)iffPis a prefix walk/suffix walk/subwalk ofQ
-
[41]
For any two open walksPandQinG,Label(P) =Label(Q)iffP=Q
-
[42]
For any vertex or edgevand closed walkPinG, the number of occurrences ofvinPis the same as the number of occurrences ofLabel(v)inLabel(P)
-
[43]
Then dis the period ofLabel(C)
LetCbe a simple circuit anddbe the total length of its edges minus total length of all vertices. Then dis the period ofLabel(C). Proof: (1)We will prove this statement only for substrings since the other cases can be proven anal- ogously. Consider the extensionQ ′ of the walkQobtained by adding one edge at the beginning and one at the end. SinceQis closed...
-
[44]
For any infix-free string-setvand chromosome-setCAG(C, V)is a non-redundant graph
-
[45]
ThenC⊆CPC(AG(C, V)), and for any other non-redundant assembly graphGsatisfyingC⊆CPC(G)andV(AG(C, V))⊆V(G), we have C⊆CPC(AG(C, V))⊆CPC(G)
LetCbe a cyclic string-set, and letVbe a proper infix-free set of strings such that every string inCcontains at least one element ofVas a substring. ThenC⊆CPC(AG(C, V)), and for any other non-redundant assembly graphGsatisfyingC⊆CPC(G)andV(AG(C, V))⊆V(G), we have C⊆CPC(AG(C, V))⊆CPC(G). Proof: (1)In non-redundant assembly graphs the number of occurrences ...
-
[46]
Eithervhas at least two outgoing prefix edges and no outgoing suffix edges or exactly one outgoing suffix edge and no outgoing prefix edges
-
[47]
3.vis unbranching (both indegree and outdegree equal to1if only if it has a maximal label by substring inclusion among all vertex labels
Eithervhas at least two incoming suffix edges and no incoming prefix edges or exactly one incoming prefix edge and no incoming suffix edges. 3.vis unbranching (both indegree and outdegree equal to1if only if it has a maximal label by substring inclusion among all vertex labels. Its incoming and outgoing edges are prefix and suffix respectively. We refer t...
-
[48]
CPC(G ∗) =CPC(G)if and only if every multiplexing step is free
-
[49]
Proof: (1)Assume thatG ∗ was obtained fromGin a single multiplexing step, but without the supre- graph condensing procedure
Any condensed supregraphG ∗ can be obtained from SPG(CPC(G∗))(minimal supregraph with the same set of chromosome candidates) by a sequence of free multiplexing steps. Proof: (1)Assume thatG ∗ was obtained fromGin a single multiplexing step, but without the supre- graph condensing procedure. Assume that the multiplexing was applied at vertexvand resulted i...
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.