Recognition: unknown
MIMIC: A Generative Multimodal Foundation Model for Biomolecules
Pith reviewed 2026-05-08 03:27 UTC · model grok-4.3
The pith
A generative model conditions on any mix of sequence, structure, evolution and regulation to reconstruct and design biomolecules.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
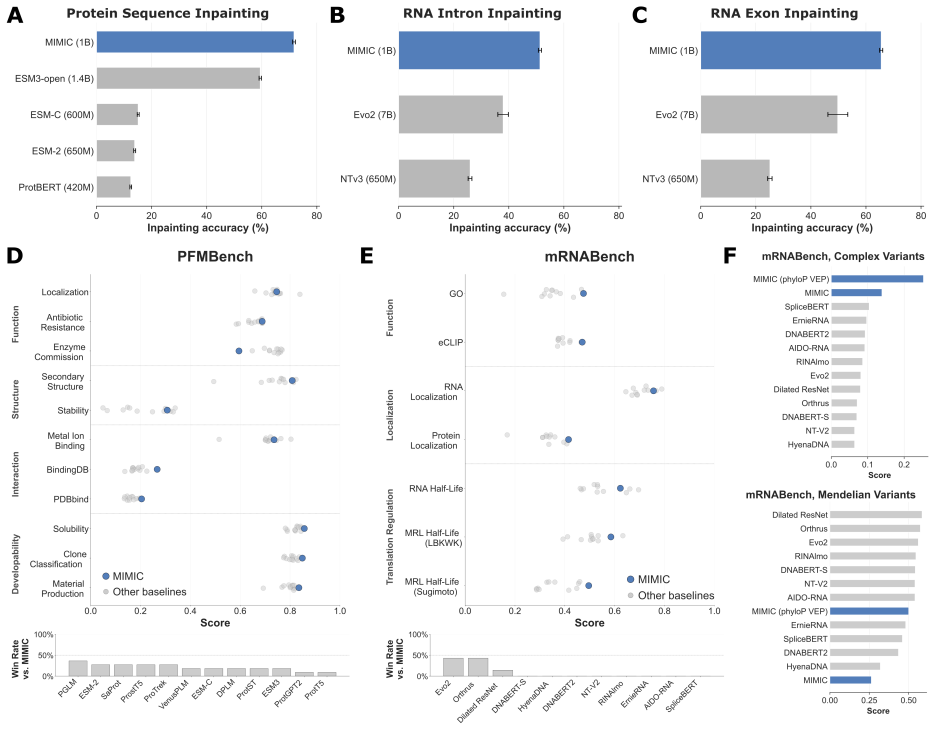
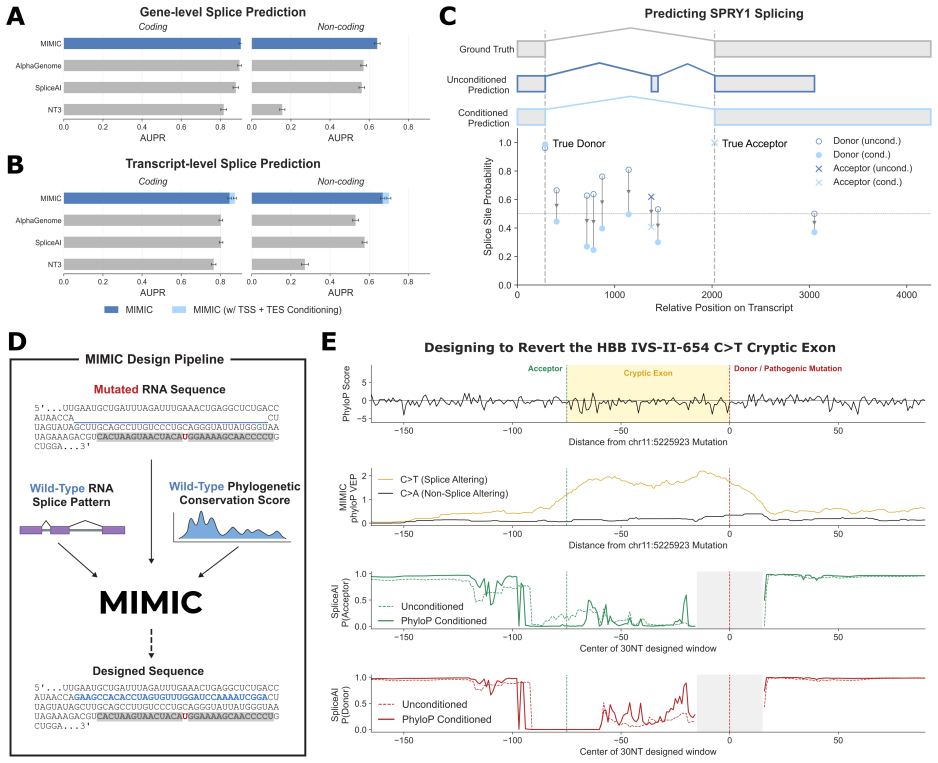
MIMIC is a generative multimodal foundation model trained on the LORE dataset linking nucleic acid, protein, evolutionary, structural, regulatory and semantic modalities within partially observed biomolecular states. The split-track encoder-decoder conditions on arbitrary subsets of observed modalities to reconstruct or generate missing components across genome, transcriptome and proteome. Multimodal conditioning improves sequence reconstruction relative to sequence-only inputs, learned representations enable state-of-the-art performance on RNA and protein downstream tasks, and the model reaches state-of-the-art splicing prediction with further gains from isoform-aware inference. The joint生成
What carries the argument
The split-track encoder-decoder architecture that processes modalities in separate tracks while sharing a joint generative space to condition on partial observations and produce complete molecular states.
If this is right
- Multimodal conditioning improves sequence reconstruction accuracy relative to sequence-only inputs.
- Learned representations enable state-of-the-art performance on RNA and protein downstream tasks.
- The model achieves state-of-the-art splicing prediction.
- Isoform-aware inference further improves performance on relevant tasks.
- The generative framework supports constrained design such as corrective RNA edits and protein sequences with target binding.
Where Pith is reading between the lines
- If the alignments learned on LORE transfer to new biomolecules or modalities, the model could support design in areas with sparse data.
- The semantic conditioning mechanism might be extended to model how experimental conditions alter molecular behavior in living cells.
- Joint modeling of evolutionary and structural signals could be tested for designing molecules that remain functional under mutation pressure.
- Applying the same split-track approach to multi-molecule complexes rather than single sequences would be a direct next test.
Load-bearing premise
The newly curated LORE dataset supplies accurate and representative alignments across nucleic acid, protein, evolutionary, structural, regulatory and semantic modalities despite many partial observations.
What would settle it
Training MIMIC on LORE and finding no consistent improvement in sequence reconstruction accuracy or downstream task performance when adding multimodal conditioning compared with sequence-only inputs on held-out test data would falsify the central claims.
Figures



read the original abstract
Biological function emerges from coupled constraints across sequence, structure, regulation, evolution, and cellular context, yet most foundation models in biology are trained within one modality or for a fixed forward task. We present MIMIC, a generative multimodal foundation model trained on our newly curated and aligned dataset, LORE, linking nucleic acid, protein, evolutionary, structural, regulatory, and semantic/contextual modalities within partially observed biomolecular states. MIMIC uses a split-track encoder-decoder architecture to condition on arbitrary subsets of observed modalities and reconstruct or generate missing components of molecular state across the genome, transcriptome, and proteome. Multimodal conditioning consistently improves MIMIC's sequence reconstruction relative to sequence-only inputs, while its learned representations enable state-of-the-art performance on RNA and protein downstream tasks. MIMIC achieves state-of-the-art splicing prediction, and its joint generative formulation enables isoform-aware inference that further improves performance. Beyond prediction, the same generative framework supports constrained design. For RNA, MIMIC identifies corrective edits in a clinically relevant HBB splice-disrupting mutation without reverting it by using evolutionary and structural signals. For proteins, jointly conditioning on shape and surface chemistry of PD-L1 and hACE2 binding sites produces diverse, high-confidence sequences with strong in silico support for target binding. Finally, MIMIC uses experimental context as semantic conditioning to model assay-dependent RNA chemical probing, rather than treating context as a fixed output. Together, these results position MIMIC's aligned multimodal generative modeling as a strong foundation for unifying representation learning, conditional prediction, and constrained biomolecular design within a single model.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. MIMIC is a generative multimodal foundation model for biomolecules trained on the newly curated LORE dataset aligning nucleic acid, protein, evolutionary, structural, regulatory, and semantic modalities under partial observations. It employs a split-track encoder-decoder architecture to condition on arbitrary modality subsets for reconstruction or generation across genome, transcriptome, and proteome. The paper claims multimodal conditioning yields consistent improvements in sequence reconstruction over sequence-only baselines, enables SOTA performance on RNA/protein downstream tasks including splicing prediction, supports isoform-aware inference, and facilitates constrained design tasks such as corrective RNA edits and protein binding sequence generation, plus context-dependent probing modeling.
Significance. If validated with quantitative evidence, the work would advance biological foundation models by providing a unified generative framework that integrates coupled constraints across modalities, potentially improving both predictive tasks and constrained design beyond single-modality approaches. The split-track handling of partial observations and isoform-aware generation represent technical strengths that could influence future multimodal modeling in genomics and proteomics.
major comments (2)
- [LORE dataset] LORE dataset section: The manuscript provides no details on alignment procedures, accuracy metrics, error rates, or controls for systematic biases from partial observations across modalities. This is load-bearing for the central claim that the split-track architecture learns useful joint representations enabling multimodal gains, as the abstract's performance assertions rest entirely on this unverified curation.
- [Abstract and results] Abstract and results summary: Claims of 'state-of-the-art splicing prediction', 'consistent improvement' in reconstruction, and 'further improves performance' via isoform-aware inference are stated without quantitative metrics, error bars, ablation studies, baseline comparisons, or dataset statistics. This prevents verification of effect sizes and undermines assessment of whether gains derive from the generative formulation or dataset artifacts.
minor comments (1)
- [Methods] The split-track architecture would benefit from an explicit diagram or pseudocode in the methods to clarify conditioning on arbitrary modality subsets.
Simulated Author's Rebuttal
We thank the referee for their constructive and detailed review, which has helped us strengthen the clarity and verifiability of our work. We have revised the manuscript to address both major comments by expanding the LORE dataset description and incorporating quantitative metrics, ablations, and comparisons throughout the abstract and results. Our point-by-point responses follow.
read point-by-point responses
-
Referee: [LORE dataset] LORE dataset section: The manuscript provides no details on alignment procedures, accuracy metrics, error rates, or controls for systematic biases from partial observations across modalities. This is load-bearing for the central claim that the split-track architecture learns useful joint representations enabling multimodal gains, as the abstract's performance assertions rest entirely on this unverified curation.
Authors: We agree that detailed documentation of the LORE dataset curation is essential to substantiate the multimodal gains and the role of the split-track architecture. In the revised manuscript we have added an expanded LORE dataset section that describes the alignment procedures (cross-referencing via stable identifiers from RefSeq, UniProt, ENCODE, and GTEx), reports accuracy metrics and error rates obtained from a held-out validation set against independent annotations, and includes controls for systematic biases (distributional comparisons, sensitivity analyses under varying partial-observation rates, and checks for annotation-source imbalances). These additions directly support that the observed improvements arise from joint representation learning rather than curation artifacts. revision: yes
-
Referee: [Abstract and results] Abstract and results summary: Claims of 'state-of-the-art splicing prediction', 'consistent improvement' in reconstruction, and 'further improves performance' via isoform-aware inference are stated without quantitative metrics, error bars, ablation studies, baseline comparisons, or dataset statistics. This prevents verification of effect sizes and undermines assessment of whether gains derive from the generative formulation or dataset artifacts.
Authors: We acknowledge that the original abstract and results summary presented claims qualitatively. We have revised both the abstract and the main results section to include the requested quantitative information: specific AUROC and accuracy values for splicing prediction with direct comparisons to prior SOTA methods, percentage improvements in sequence reconstruction together with standard deviations across multiple runs, ablation tables isolating each modality and the isoform-aware component, baseline comparisons on the same datasets, and summary statistics of the LORE splits. These additions allow verification of effect sizes and confirm that the gains are attributable to the multimodal generative formulation. revision: yes
Circularity Check
No significant circularity; empirical claims rest on external benchmarks and held-out evaluations rather than self-referential inputs.
full rationale
The paper's core claims concern empirical improvements from multimodal conditioning on the LORE dataset and SOTA results on RNA/protein downstream tasks. No equations, derivations, or self-citations are presented that reduce any prediction to its own fitted inputs by construction. The split-track architecture and generative formulation are described as standard encoder-decoder components conditioned on observed modalities, with performance gains asserted via comparisons to sequence-only baselines and prior models. These evaluations are positioned against external tasks and benchmarks, rendering the work self-contained. While the newly curated LORE dataset introduces a potential verification gap regarding alignment accuracy, this is an empirical concern rather than a circular reduction in the derivation chain. No self-definitional, fitted-input-renamed-as-prediction, or load-bearing self-citation patterns are identifiable from the provided text.
Axiom & Free-Parameter Ledger
free parameters (2)
- LORE dataset alignment parameters
- Model hyperparameters
axioms (1)
- domain assumption Partially observed biomolecular states can be reconstructed or generated from arbitrary subsets of modalities using a shared latent space.
invented entities (1)
-
LORE dataset
no independent evidence
Reference graph
Works this paper leans on
-
[1]
Francis H. C. Crick. Central dogma of molecular biology.Nature, 227(5258):561–563, 1970
1970
-
[2]
Integrative analysis of 111 reference human epigenomes
Roadmap Epigenomics Consortium. Integrative analysis of 111 reference human epigenomes. Nature, 518(7539):317–330, 2015
2015
-
[3]
Expanded encyclopaedias of dna elements in the human and mouse genomes.Nature, 583(7818):699–710, 2020
ENCODE Project Consortium. Expanded encyclopaedias of dna elements in the human and mouse genomes.Nature, 583(7818):699–710, 2020
2020
-
[4]
Buenrostro, Paul G
Jason D. Buenrostro, Paul G. Giresi, Lisa C. Zaba, Howard Y. Chang, and William J. Green- leaf. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, dna-binding proteins and nucleosome position.Nature Methods, 10(12):1213–1218, 2013
2013
-
[5]
Anfinsen
Christian B. Anfinsen. Principles that govern the folding of protein chains.Science, 181(4096):223–230, 1973
1973
-
[6]
Ulrich Hartl, Andreas Bracher, and Manajit Hayer-Hartl
F. Ulrich Hartl, Andreas Bracher, and Manajit Hayer-Hartl. Molecular chaperones in protein folding and proteostasis.Nature, 475(7356):324–332, 2011
2011
-
[7]
Pedersen, Angie S
Adam Siepel, Gill Bejerano, Jakob S. Pedersen, Angie S. Hinrichs, Minmei Hou, Kate Rosen- bloom, Hiram Clawson, John Spieth, LaDeana W. Hillier, Stephen Richards, George M. We- instock, Richard K. Wilson, Richard A. Gibbs, W. James Kent, Webb Miller, and David Haussler. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Geno...
2005
-
[9]
Davydov, Daniel L
Eugene V. Davydov, Daniel L. Goode, Marina Sirota, Gregory M. Cooper, Arend Sidow, and Serafim Batzoglou. Identifying a high fraction of the human genome to be under selective constraint using GERP++.PLoS Computational Biology, 6(12):e1001025, 2010
2010
-
[10]
Biobert: a pre-trained biomedical language representation model for biomedical text mining.Bioinformatics, 36(4):1234–1240, 2020
Jinhyuk Lee, Wonjin Yoon, Sungdong Kim, Donghyeon Kim, Sunkyu Kim, Chan Ho So, and Jaewoo Kang. Biobert: a pre-trained biomedical language representation model for biomedical text mining.Bioinformatics, 36(4):1234–1240, 2020
2020
-
[11]
Towards generalist biomedical ai.Nejm Ai, 1(3):AIoa2300138, 2024
Tao Tu, Shekoofeh Azizi, Danny Driess, Mike Schaekermann, Mohamed Amin, Pi-Chuan Chang, Andrew Carroll, Charles Lau, Ryutaro Tanno, Ira Ktena, et al. Towards generalist biomedical ai.Nejm Ai, 1(3):AIoa2300138, 2024
2024
-
[12]
Maddison, and Bo Wang
AdibvafaFallahpour, AndrewMagnuson, PuravGupta, ShihaoMa, JackNaimer, ArnavShah, Haonan Duan, Omar Ibrahim, Hani Goodarzi, Chris J. Maddison, and Bo Wang. Bioreason: Incentivizing multimodal biological reasoning within a dna-llm model, 2025
2025
-
[13]
Prottex: Structure-in-context reasoning and editing of proteins with large language models, 2025
Zicheng Ma, Chuanliu Fan, Zhicong Wang, Zhenyu Chen, Xiaohan Lin, Yanheng Li, Shihao Feng, Jun Zhang, Ziqiang Cao, and Yi Qin Gao. Prottex: Structure-in-context reasoning and editing of proteins with large language models, 2025. 18
2025
-
[14]
Domain-specific language model pretraining for biomedical natural language processing.ACM Trans
Yu Gu, Robert Tinn, Hao Cheng, Michael Lucas, Naoto Usuyama, Xiaodong Liu, Tristan Naumann, Jianfeng Gao, and Hoifung Poon. Domain-specific language model pretraining for biomedical natural language processing.ACM Trans. Comput. Healthcare, 3(1), October 2021
2021
-
[15]
Sara Mahdavi, Christopher Semturs, David Fleet, Vivek Natarajan, and Shekoofeh Azizi
Juan Manuel Zambrano Chaves, Eric Wang, Tao Tu, Eeshit Dhaval Vaishnav, Byron Lee, S. Sara Mahdavi, Christopher Semturs, David Fleet, Vivek Natarajan, and Shekoofeh Azizi. Tx-llm: A large language model for therapeutics, 2024
2024
-
[16]
Biogpt: generative pre-trained transformer for biomedical text generation and mining
Renqian Luo, Liai Sun, Yingce Xia, Tao Qin, Sheng Zhang, Hoifung Poon, and Tie-Yan Liu. Biogpt: generative pre-trained transformer for biomedical text generation and mining. Briefings in Bioinformatics, 23(6), 9 2022
2022
-
[17]
Biomistral: A collection of open-source pretrained large language models for medical domains, 2024
Yanis Labrak, Adrien Bazoge, Emmanuel Morin, Pierre-Antoine Gourraud, Mickael Rouvier, and Richard Dufour. Biomistral: A collection of open-source pretrained large language models for medical domains, 2024
2024
-
[18]
Tristan Bepler and Bonnie Berger. Learning protein sequence embeddings using information from structure.arXiv preprint arXiv:1902.08661, 2019
-
[19]
Unified rational protein engineering with sequence-based deep representation learn- ing.Nature methods, 16(12):1315–1322, 2019
Ethan C Alley, Grigory Khimulya, Surojit Biswas, Mohammed AlQuraishi, and George M Church. Unified rational protein engineering with sequence-based deep representation learn- ing.Nature methods, 16(12):1315–1322, 2019
2019
-
[20]
Alexander Rives, Joshua Meier, Tom Sercu, Siddharth Goyal, Zeming Lin, Jason Liu, Demi Guo, Myle Ott, C Lawrence Zitnick, Jerry Ma, et al. Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences.Proceedings of the national academy of sciences, 118(15):e2016239118, 2021
2021
-
[21]
Evolutionary-scale prediction of atomic- level protein structure with a language model.Science, 379(6637):1123–1130, 2023
Zeming Lin, Halil Akin, Roshan Rao, Brian Hie, Zhongkai Zhu, Wenting Lu, Nikita Smetanin, Robert Verkuil, Ori Kabeli, Yaniv Shmueli, Allan dos Santos Costa, Maryam Fazel-Zarandi, Tom Sercu, Salvatore Candido, and Alexander Rives. Evolutionary-scale prediction of atomic- level protein structure with a language model.Science, 379(6637):1123–1130, 2023
2023
-
[22]
Bo Chen, Xingyi Cheng, Pan Li, Yangli-ao Geng, Jing Gong, Shen Li, Zhilei Bei, Xu Tan, Boyan Wang, Xin Zeng, et al. xtrimopglm: unified 100b-scale pre-trained transformer for deciphering the language of protein.arXiv preprint arXiv:2401.06199, 2024
-
[23]
Prottrans: toward understanding the language of life through self-supervised learning.IEEE transactions on pattern analysis and machine intelligence, 44(10):7112–7127, 2021
Ahmed Elnaggar, Michael Heinzinger, Christian Dallago, Ghalia Rehawi, Yu Wang, Llion Jones, Tom Gibbs, Tamas Feher, Christoph Angerer, Martin Steinegger, et al. Prottrans: toward understanding the language of life through self-supervised learning.IEEE transactions on pattern analysis and machine intelligence, 44(10):7112–7127, 2021
2021
-
[24]
Poet: A generative model of protein families as sequences-of-sequences.Advances in Neural Information Processing Systems, 36:77379–77415, 2023
Timothy Truong Jr and Tristan Bepler. Poet: A generative model of protein families as sequences-of-sequences.Advances in Neural Information Processing Systems, 36:77379–77415, 2023
2023
-
[25]
Simulating 500 million years of evolution with a language model.Science, 2025
Thomas Hayes, Roshan Rao, Halil Akin, et al. Simulating 500 million years of evolution with a language model.Science, 2025
2025
-
[26]
Timothy Fei Truong Jr and Tristan Bepler. Understanding protein function with a multimodal retrieval-augmented foundation model.arXiv preprint arXiv:2508.04724, 2025. 19
-
[27]
A trimodal protein language model enables advanced protein searches.Nature Biotechnology, pages 1–7, 2025
Jin Su, Yan He, Shiyang You, Shiyu Jiang, Xibin Zhou, Xuting Zhang, Yuxuan Wang, Xining Su, Igor Tolstoy, Xing Chang, et al. A trimodal protein language model enables advanced protein searches.Nature Biotechnology, pages 1–7, 2025
2025
-
[28]
Protst: Multi-modality learning of protein sequences and biomedical texts
Minghao Xu, Xinyu Yuan, Santiago Miret, and Jian Tang. Protst: Multi-modality learning of protein sequences and biomedical texts. InInternational Conference on Machine Learning, pages 38749–38767. PMLR, 2023
2023
-
[29]
Durrant, Jerome Ku, Michael Poli, et al
Garyk Brixi, Matthew G. Durrant, Jerome Ku, Michael Poli, et al. Genome modeling and design across all domains of life with Evo 2.bioRxiv, 2025
2025
-
[30]
Koo, Alexander Stark, Volodymyr Kuleshov, et al
Juan Boshar, Richard Evans, Thomas Pierrot, Peter K. Koo, Alexander Stark, Volodymyr Kuleshov, et al. A foundational model for joint sequence-function multi-species modeling at scale for long-range genomic prediction.bioRxiv, 2025
2025
-
[31]
Durrant, et al
Eric Nguyen, Michael Poli, Matthew G. Durrant, et al. HyenaDNA: Long-range genomic sequence modeling at single nucleotide resolution, 2023. NeurIPS 2023
2023
-
[32]
Caduceus: Bi-directional equivariant long-range DNA sequence modeling, 2024
Yair Schiff, Chia-Hsiang Kao, Aaron Gokaslan, Tri Dao, Albert Gu, and Volodymyr Kuleshov. Caduceus: Bi-directional equivariant long-range DNA sequence modeling, 2024
2024
-
[33]
DNABERT-2: Efficient foundation model and benchmark for multi-species genome, 2023
Zhihan Zhou, Yanrong Ji, Weijian Li, Pratik Dutta, Ramana Davuluri, and Han Liu. DNABERT-2: Efficient foundation model and benchmark for multi-species genome, 2023
2023
-
[34]
Ledsam, Agnieszka Grabska- Barwinska, Kyle R
Žiga Avsec, Vikram Agarwal, Daniel Visentin, Joseph R. Ledsam, Agnieszka Grabska- Barwinska, Kyle R. Taylor, Yannis Assael, et al. Effective gene expression prediction from sequence by integrating long-range interactions.Nature Methods, 18(10):1196–1203, 2021
2021
-
[35]
Advancing regulatory variant effect prediction with AlphaGenome.Nature, 649(8099):1206–1218, 2026
Žiga Avsec et al. Advancing regulatory variant effect prediction with AlphaGenome.Nature, 649(8099):1206–1218, 2026
2026
-
[36]
Kelley, Yakir A
David R. Kelley, Yakir A. Reshef, Maxwell Bileschi, David Belanger, Cory Y. McLean, and JasperSnoek. Sequentialregulatoryactivitypredictionacrosschromosomeswithconvolutional neural networks.Genome Research, 28(5):739–750, 2018
2018
-
[37]
Troyanskaya
Jian Zhou and Olga G. Troyanskaya. Predicting effects of noncoding variants with deep learning–based sequence model.Nature Methods, 12(10):931–934, 2015
2015
-
[38]
Chen, Aaron K
Kathleen M. Chen, Aaron K. Wong, et al. A sequence-based global map of regulatory activity for deciphering human genetics.Nature Genetics, 54(7):940–949, 2022
2022
-
[39]
Johannes Linder, Divyanshi Srivastava, Han Yuan, Vikram Agarwal, and David R. Kelley. Predicting RNA-seq coverage from DNA sequence as a unifying model of gene regulation. Nature Genetics, 57(4):949–961, 2025
2025
-
[40]
David R. Kelley. Cross-species regulatory sequence activity prediction.PLoS Computational Biology, 16(7):e1008050, 2020
2020
-
[41]
An interpretable rna foundation model for exploring functional rna motifs in plants.Nature Machine Intelligence, 6(12):1616–1625, 2024
Haopeng Yu, Heng Yang, Wenqing Sun, Zongyun Yan, Xiaofei Yang, Huakun Zhang, Yiliang Ding, and Ke Li. An interpretable rna foundation model for exploring functional rna motifs in plants.Nature Machine Intelligence, 6(12):1616–1625, 2024. 20
2024
-
[42]
Machine learning a model for rna structure prediction.NAR genomics and bioinformatics, 2(4):lqaa090, 2020
Nicola Calonaci, Alisha Jones, Francesca Cuturello, Michael Sattler, and Giovanni Bussi. Machine learning a model for rna structure prediction.NAR genomics and bioinformatics, 2(4):lqaa090, 2020
2020
-
[43]
Accurate rna 3d structure prediction using a language model-based deep learning approach.Nature Methods, 21(12):2287–2298, 2024
Tao Shen, Zhihang Hu, Siqi Sun, Di Liu, Felix Wong, Jiuming Wang, Jiayang Chen, Yixuan Wang, Liang Hong, Jin Xiao, et al. Accurate rna 3d structure prediction using a language model-based deep learning approach.Nature Methods, 21(12):2287–2298, 2024
2024
-
[44]
Geometric deep learning of rna structure.Science, 373(6558):1047–1051, 2021
Raphael JL Townshend, Stephan Eismann, Andrew M Watkins, Ramya Rangan, Masha Karelina, Rhiju Das, and Ron O Dror. Geometric deep learning of rna structure.Science, 373(6558):1047–1051, 2021
2021
-
[45]
Machine learning for rna 2d structure prediction benchmarked on experimental data.Briefings in Bioinformatics, 24(3):bbad153, 2023
Marek Justyna, Maciej Antczak, and Marta Szachniuk. Machine learning for rna 2d structure prediction benchmarked on experimental data.Briefings in Bioinformatics, 24(3):bbad153, 2023
2023
-
[46]
Highly accurate protein structure prediction with AlphaFold.Nature, 596(7873):583–589, 2021
John Jumper, Richard Evans, Alexander Pritzel, et al. Highly accurate protein structure prediction with AlphaFold.Nature, 596(7873):583–589, 2021
2021
-
[47]
Accurate prediction of protein structures and interactions using a three-track neural network.Science, 373(6557):871–876, 2021
Minkyung Baek, Frank DiMaio, Ivan Anishchenko, et al. Accurate prediction of protein structures and interactions using a three-track neural network.Science, 373(6557):871–876, 2021
2021
-
[48]
Accuratestructurepredictionofbiomolecular interactions with AlphaFold 3.Nature, 630(8016):493–500, 2024
JoshAbramson, JonasAdler, JackDunger, etal. Accuratestructurepredictionofbiomolecular interactions with AlphaFold 3.Nature, 630(8016):493–500, 2024
2024
-
[49]
High-resolution de novo structure prediction from primary sequence.BioRxiv, pages 2022–07, 2022
Ruidong Wu, Fan Ding, Rui Wang, Rui Shen, Xiwen Zhang, Shitong Luo, Chenpeng Su, Zuofan Wu, Qi Xie, Bonnie Berger, et al. High-resolution de novo structure prediction from primary sequence.BioRxiv, pages 2022–07, 2022
2022
-
[50]
McRae, Shawn F
Kishore Jaganathan, Sofia Kyriazopoulou Panagiotopoulou, James F. McRae, Shawn F. Dar- bandi, David Knowles, Yang I. Li, Jack A. Kosmicki, Juan Arbelaez, Wei Cui, Guy B. Schwartz, Eric D. Chow, Elias Kanterakis, Han Gao, Amir Kia, Serafim Batzoglou, Stephan J. Sanders, and Kyle K.-H. Farh. Predicting splicing from primary sequence with deep learning. Cell...
2019
-
[51]
Tony Zeng and Yang I. Li. Predicting RNA splicing from DNA sequence using Pangolin. Genome Biology, 23(1):103, 2022
2022
-
[52]
Variant-resolved predic- tion of context-specific isoform variation with a graph-based attention model.Cell Genomics, 2026
Aviya Litman, Zhicheng Pan, Ksenia Sokolova, Joyce Fang, Tess Marvin, Natalie Sauerwald, Christopher Y Park, Chandra L Theesfeld, and Olga G Troyanskaya. Variant-resolved predic- tion of context-specific isoform variation with a graph-based attention model.Cell Genomics, 2026
2026
-
[53]
4M: Massively multimodal masked modeling
David Mizrahi, Roman Bachmann, Oğuzhan Fatih Kar, Teresa Yeo, Mingfei Gao, Afshin Dehghan, and Amir Zamir. 4M: Massively multimodal masked modeling. InThirty-seventh Conference on Neural Information Processing Systems, 2023
2023
-
[54]
AION-1: Omnimodal foundation model for astronomical sciences, 2025
Liam Parker, Francois Lanusse, Jeff Shen, Ollie Liu, Tom Hehir, Leopoldo Sarra, Lucas Meyer, Micah Bowles, Sebastian Wagner-Carena, Helen Qu, Siavash Golkar, Alberto Bietti, Hatim Bourfoune, Nathan Casserau, Pierre Cornette, Keiya Hirashima, Geraud Krawezik, Ruben Ohana, Nicholas Lourie, Michael McCabe, Rudy Morel, Payel Mukhopadhyay, Mariel Pettee, 21 Br...
2025
-
[55]
OpenGenome2: a database of nearly 9 trillion base pairs of curated DNA from across all domains of life
Arc Institute. OpenGenome2: a database of nearly 9 trillion base pairs of curated DNA from across all domains of life. Hugging Face dataset, 2025. Dataset: arcinstitute/opengenome2
2025
-
[56]
Clustering huge protein sequence sets in linear time
Martin Steinegger and Johannes Söding. Clustering huge protein sequence sets in linear time. Nature Communications, 9(1):2542, 2018
2018
-
[57]
MMseqs2enablessensitiveproteinsequencesearching for the analysis of massive data sets.Nature Biotechnology, 35(11):1026–1028, 2017
MartinSteineggerandJohannesSöding. MMseqs2enablessensitiveproteinsequencesearching for the analysis of massive data sets.Nature Biotechnology, 35(11):1026–1028, 2017
2017
-
[58]
O’Leary et al
Nuala A. O’Leary et al. Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation.Nucleic Acids Research, 44(D1):D733–D745, 2016
2016
-
[59]
Mudge et al
Jonathan M. Mudge et al. GENCODE 2025: reference gene annotation for human and mouse. Nucleic Acids Research, 53(D1):D966–D975, 2025
2025
-
[60]
Rasp v2.0: an updated atlas for rna structure probing data.Nucleic Acids Research, 53(D1):D211–D219, 11 2025
Kunting Mu, Yuhan Fei, Yiran Xu, and Qiangfeng Cliff Zhang. Rasp v2.0: an updated atlas for rna structure probing data.Nucleic Acids Research, 53(D1):D211–D219, 11 2025
2025
-
[61]
CAGE: cap analysis of gene expression.Nature Methods, 3(3):211– 222, 2006
Rimantas Kodzius et al. CAGE: cap analysis of gene expression.Nature Methods, 3(3):211– 222, 2006
2006
-
[62]
UniProt: the universal protein knowledgebase in 2025.Nucleic Acids Research, 53(D1):D609–D617, 2025
The UniProt Consortium. UniProt: the universal protein knowledgebase in 2025.Nucleic Acids Research, 53(D1):D609–D617, 2025
2025
-
[63]
Mihaly Varadi et al. Alphafold protein structure database: massively expanding the struc- tural coverage of protein-sequence space with high-accuracy models.Nucleic Acids Research, 50(D1):D439–D444, 2022
2022
-
[64]
Deciphering interaction fingerprints from protein molecular surfaces using geometric deep learning.Nature Methods, 17(2):184–192, 2020
Pablo Gainza et al. Deciphering interaction fingerprints from protein molecular surfaces using geometric deep learning.Nature Methods, 17(2):184–192, 2020
2020
-
[65]
UniProtKB/Swiss-Prot.Methods in Molecular Biology (Clifton, N.J.), 406:89–112, 2007
Emmanuel Boutet, Damien Lieberherr, Michael Tognolli, Michel Schneider, and Amos Bairoch. UniProtKB/Swiss-Prot.Methods in Molecular Biology (Clifton, N.J.), 406:89–112, 2007
2007
-
[66]
Paxdb 5.0: curated protein quantification data suggests adaptive proteome changes in yeasts.Molecular & Cellular Proteomics, 22(10), 2023
Qingyao Huang, Damian Szklarczyk, Mingcong Wang, Milan Simonovic, and Christian von Mering. Paxdb 5.0: curated protein quantification data suggests adaptive proteome changes in yeasts.Molecular & Cellular Proteomics, 22(10), 2023
2023
-
[67]
Democratizing protein language models with parameter-efficient fine- tuning.Proceedings of the National Academy of Sciences, 121(26):e2405840121, 2024
Samuel Sledzieski, Meghana Kshirsagar, Minkyung Baek, Rahul Dodhia, Juan Lavista Ferres, and Bonnie Berger. Democratizing protein language models with parameter-efficient fine- tuning.Proceedings of the National Academy of Sciences, 121(26):e2405840121, 2024
2024
-
[68]
Fine-tuning protein language models boosts predictions across diverse tasks.Nature Communications, 15(1):7407, 2024
Robert Schmirler, Michael Heinzinger, and Burkhard Rost. Fine-tuning protein language models boosts predictions across diverse tasks.Nature Communications, 15(1):7407, 2024
2024
-
[69]
Lora: Low-rank adaptation of large language models.Iclr, 1(2):3, 2022
Edward J Hu, Yelong Shen, Phillip Wallis, Zeyuan Allen-Zhu, Yuanzhi Li, Shean Wang, Liang Wang, Weizhu Chen, et al. Lora: Low-rank adaptation of large language models.Iclr, 1(2):3, 2022. 22
2022
-
[70]
Pfmbench: Protein foundation model benchmark.arXiv preprint arXiv:2506.14796, 2025
Zhangyang Gao, Hao Wang, Cheng Tan, Chenrui Xu, Mengdi Liu, Bozhen Hu, Linlin Chao, Xiaoming Zhang, and Stan Z Li. Pfmbench: Protein foundation model benchmark.arXiv preprint arXiv:2506.14796, 2025
-
[71]
Laverty, Ilyes Baali, Bo Wang, and Quaid Morris
Ruian Shi, Taykhoom Dalal, Philip Fradkin, Divya Koyyalagunta, Simran Chhabria, Andrew Jung, Cyrus Tam, Defne Ceyhan, Jessica Lin, Kaitlin U. Laverty, Ilyes Baali, Bo Wang, and Quaid Morris. mrnabench: A curated benchmark for mature mrna property and function prediction.bioRxiv, 2025
2025
-
[72]
Esm cambrian: Revealing the mysteries of proteins with unsupervised learning, 2024
ESM Team. Esm cambrian: Revealing the mysteries of proteins with unsupervised learning, 2024
2024
-
[73]
SaProt: Pro- tein Language Modeling with Structure-aware Vocabulary.bioRxiv, page 2023.10.01.560349, 2023
Jin Su, Chenchen Han, Yuyang Zhou, Junjie Shan, Xibin Zhou, and Fajie Yuan. SaProt: Pro- tein Language Modeling with Structure-aware Vocabulary.bioRxiv, page 2023.10.01.560349, 2023
2023
-
[74]
Orthrus: towards evolutionary and functional rna foundation models
Philip Fradkin, Ruian Shi, Taykhoom Dalal, Keren Isaev, Brendan J Frey, Leo J Lee, Quaid Morris, and Bo Wang. Orthrus: towards evolutionary and functional rna foundation models. BioRxiv, pages 2024–10, 2025
2024
-
[75]
Isoform-resolved mrna profiling of ribosome load defines interplay of hif and mtor dysregulation in kidney cancer.Nature structural & molecular biology, 29(9):871–880, 2022
Yoichiro Sugimoto and Peter J Ratcliffe. Isoform-resolved mrna profiling of ribosome load defines interplay of hif and mtor dysregulation in kidney cancer.Nature structural & molecular biology, 29(9):871–880, 2022
2022
-
[76]
Combinatorial optimization of mrna structure, stability, and translation for rna-based thera- peutics.Nature communications, 13(1):1536, 2022
Kathrin Leppek, Gun Woo Byeon, Wipapat Kladwang, Hannah K Wayment-Steele, Craig H Kerr, Adele F Xu, Do Soon Kim, Ved V Topkar, Christian Choe, Daphna Rothschild, et al. Combinatorial optimization of mrna structure, stability, and translation for rna-based thera- peutics.Nature communications, 13(1):1536, 2022
2022
-
[77]
Self- supervised learning on millions of primary rna sequences from 72 vertebrates improves sequence-based rna splicing prediction.Briefings in bioinformatics, 25(3):bbae163, 2024
Ken Chen, Yue Zhou, Maolin Ding, Yu Wang, Zhixiang Ren, and Yuedong Yang. Self- supervised learning on millions of primary rna sequences from 72 vertebrates improves sequence-based rna splicing prediction.Briefings in bioinformatics, 25(3):bbae163, 2024
2024
-
[78]
Regulation of pre-mrna splic- ing: roles in physiology and disease, and therapeutic prospects.Nature Reviews Genetics, 24(4):251–269, 2023
Malgorzata Ewa Rogalska, Claudia Vivori, and Juan Valcárcel. Regulation of pre-mrna splic- ing: roles in physiology and disease, and therapeutic prospects.Nature Reviews Genetics, 24(4):251–269, 2023
2023
-
[79]
Strauch, J
Y. Strauch, J. Lord, M. Niranjan, and D. Baralle. Ci-spliceai—improving machine learning predictions of disease causing splicing variants using curated alternative splice sites.PLOS ONE, 17(6):e0269159, 2022
2022
-
[80]
Natan Belchikov, Justine Hsu, Xiang Jennie Li, Julien Jarroux, Wen Hu, Anoushka Joglekar, and Hagen U. Tilgner. Understanding isoform expression by pairing long-read sequencing with single-cell and spatial transcriptomics.Genome Research, 34(11):1735–1746, 2024
2024
-
[81]
Steinmetz
Chenchen Zhu, Jingyan Wu, Han Sun, Francesca Briganti, Benjamin Meder, Wu Wei, and Lars M. Steinmetz. Single-molecule, full-length transcript isoform sequencing reveals disease- associated rna isoforms in cardiomyocytes.Nature Communications, 12:4203, 2021
2021
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.