Recognition: unknown
Enhancing molecular dynamics with equivariant machine-learned densities
Pith reviewed 2026-05-07 17:41 UTC · model grok-4.3
The pith
Learning the electron density first with equivariant networks enables molecular dynamics trajectories that also predict spectroscopic observables like infrared spectra.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
DenSNet learns the Hohenberg-Kohn map from nuclear configurations to the ground-state electron density via an SE(3)-equivariant neural network that outputs coefficients in a flexible atom-centered Gaussian basis, applies a delta-learning strategy with superposed atomic densities as prior, and uses a second equivariant network to map the predicted density to total energy, thereby supplying both forces for dynamics and the density for electronic observables.
What carries the argument
An SE(3)-equivariant neural network that predicts density coefficients of an atom-centered Gaussian basis, augmented by delta-learning from superposed atomic densities as a prior, followed by a second network that converts the density into total energy.
If this is right
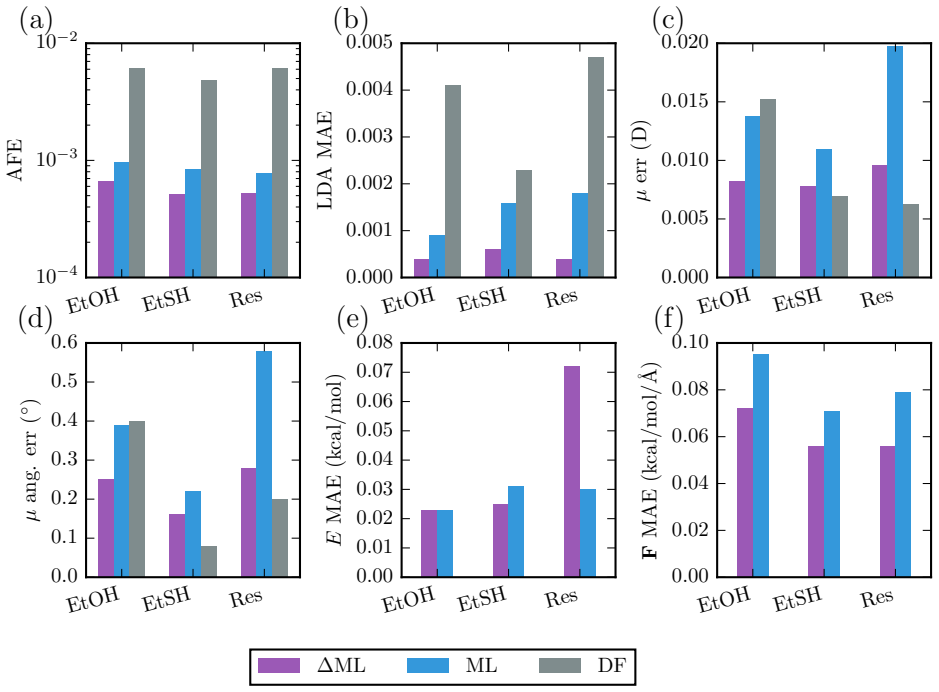
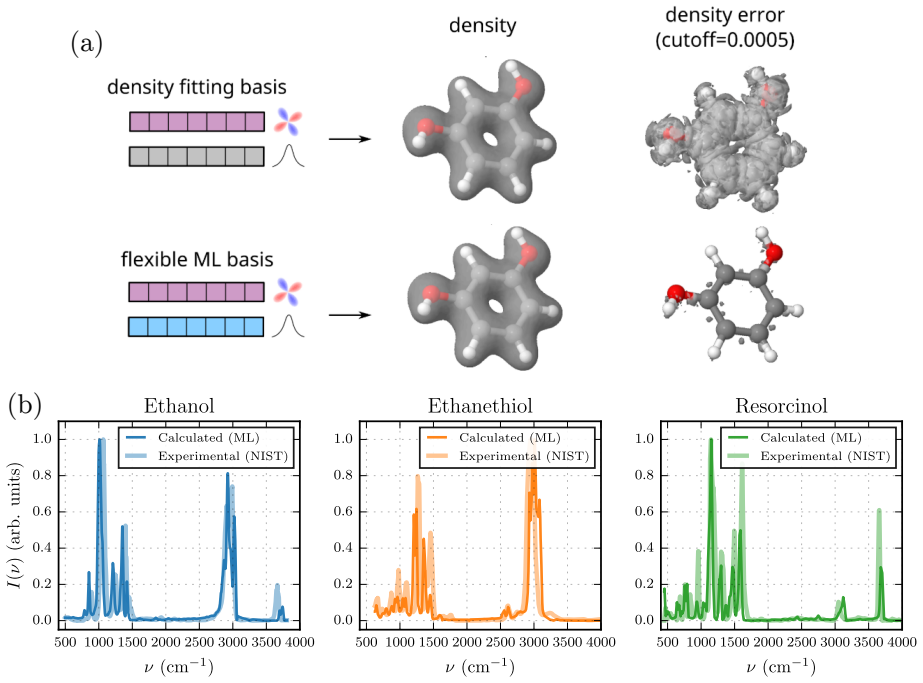
- Machine-learned trajectories on ethanol, ethanethiol, and resorcinol yield infrared spectra in excellent agreement with experimental gas-phase measurements.
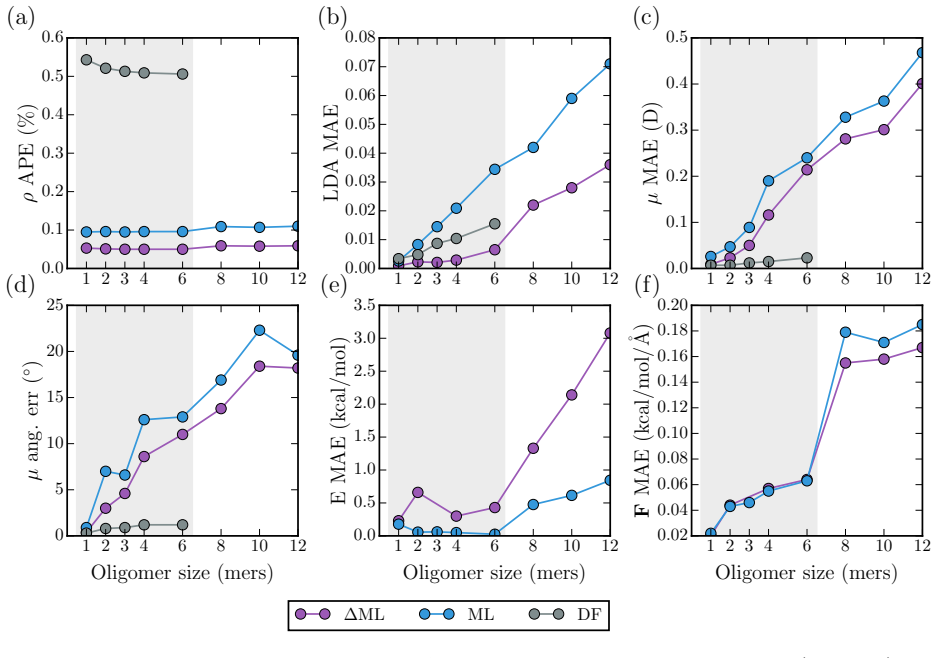
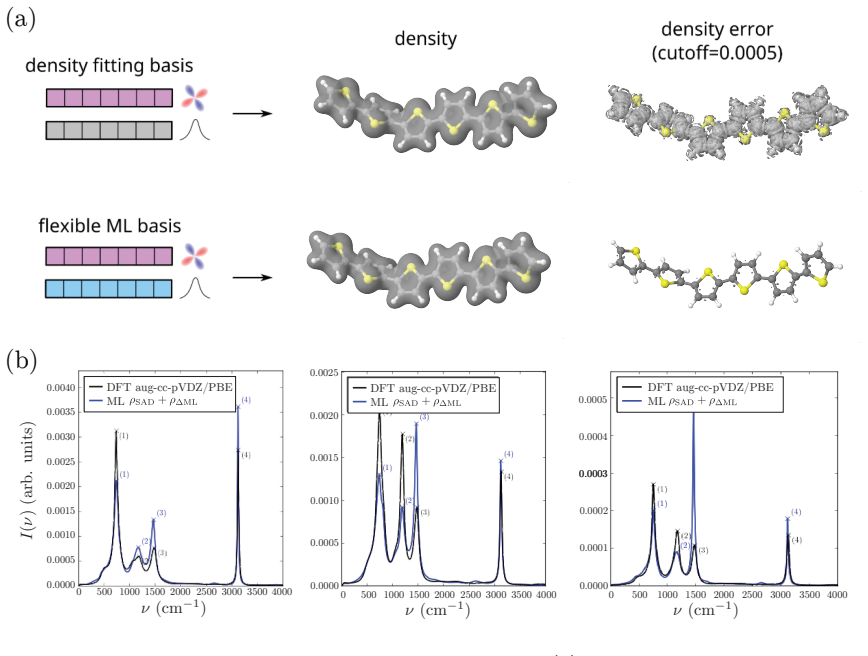
- Training on polythiophene oligomers of 1-6 monomers produces stable trajectories up to 12 monomers whose infrared spectra match reference density functional theory calculations.
- The density-first framework supplies a single model for both interatomic forces and electronic observables without separate post-processing networks.
- The approach supports extrapolation in system size while preserving transferability of spectroscopic predictions.
Where Pith is reading between the lines
- The same density representation could be used to compute additional observables such as polarizabilities or NMR shifts by adding simple post-processing operators.
- Because the density is learned directly, the model may transfer more readily across chemical composition than energy-only potentials when the underlying density functional is fixed.
- Extending the Gaussian basis or the equivariant architecture could enable simulations of larger polymers or solvated systems while retaining electronic information.
Load-bearing premise
The predicted densities remain accurate and stable over long trajectories so that errors in energies, forces, and derived observables do not accumulate and invalidate the dynamics or spectra.
What would settle it
A long molecular-dynamics trajectory driven by the learned model in which the infrared spectrum extracted from the trajectory deviates substantially from the spectrum obtained from reference density-functional-theory dynamics on the same system.
Figures




read the original abstract
Machine-learning interatomic potentials (MLIPs) have enabled molecular dynamics at near ab initio accuracy, yet remain limited to energies and forces by construction, leaving electronic observables such as dipole moments and polarizabilities inaccessible. We introduce DenSNet, a density-first approach to machine-learned electronic structure that learns the Hohenberg--Kohn map from nuclear configurations to the ground-state electron density. Our approach employs an SE(3)-equivariant neural network to predict density coefficients of a flexible atom-centered Gaussian basis, combined with a $\Delta$-learning strategy that uses superposed atomic densities as a prior to accelerate training. A second equivariant network then maps the predicted density to the total energy, providing a unified framework for molecular dynamics and electronic structure. We validate DenSNet on ethanol, ethanethiol, and resorcinol, where infrared spectra from machine-learned trajectories show excellent agreement with experimental gas-phase measurements. To test scalability, we train on polythiophene oligomers with 1--6 monomers and extrapolate to chains of up to 12 monomers, generating stable long-time trajectories whose infrared spectra agree with reference density functional theory calculations. Here, we show that reinstating the electron density as the central learned quantity opens a practical route to transferable prediction of spectroscopic and electronic observables in large-scale molecular simulations.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript introduces DenSNet, an SE(3)-equivariant neural network framework that learns the ground-state electron density from nuclear configurations via a flexible atom-centered Gaussian basis and a delta-learning strategy using superposed atomic densities as prior. A second equivariant network then maps the predicted density to total energy, enabling unified molecular dynamics and access to electronic observables such as dipole moments and infrared spectra. Validation on ethanol, ethanethiol, and resorcinol shows IR spectra from ML trajectories in good agreement with gas-phase experiments; scalability is tested by training on polythiophene oligomers (1-6 monomers) and extrapolating to 12-monomer chains, where trajectories remain stable and IR spectra match reference DFT.
Significance. If the central claims hold, the work provides a density-centric route to transferable ML potentials that simultaneously deliver spectroscopic observables without separate post-processing models. The equivariant architecture and physically motivated atomic-density prior are strengths that could improve data efficiency and symmetry preservation compared to direct energy/force fitting. The reported extrapolation to longer oligomers and experimental spectral agreement suggest practical utility for large-scale simulations, though this depends on unquantified aspects of density and force accuracy.
major comments (3)
- [Abstract] Abstract: the claim of 'stable long-time trajectories' and 'excellent agreement' for extrapolated 12-monomer IR spectra is not supported by any reported quantitative metrics on density coefficient errors, force MAEs, or energy conservation (e.g., drift per ps). Because forces are obtained by chain-rule differentiation through the predicted density coefficients, residual density inaccuracies can be amplified; without these diagnostics the weakest assumption (density stability over long trajectories) remains untested and load-bearing for the transferable-dynamics claim.
- [Abstract] The composite architecture (density NN + density-to-energy NN) is presented as unified, yet no ablation or error-propagation analysis is given showing that density prediction residuals do not degrade force accuracy relative to direct energy/force MLIPs. This is central because the paper's strongest claim is that reinstating density as the learned quantity enables reliable observables and dynamics.
- [Abstract] Validation on polythiophene extrapolation: while spectral agreement with DFT is stated, the manuscript provides no out-of-distribution force or energy error statistics on the 7-12 monomer chains, nor training-set size or composition details. This leaves the transferability assertion only partially supported.
minor comments (2)
- The abstract and text would benefit from explicit statements of the Gaussian basis size, number of density coefficients per atom, and training-set sizes for both the density and energy networks.
- Notation for the delta-learning prior and the precise form of the atom-centered Gaussian expansion should be defined with an equation in the methods section for reproducibility.
Simulated Author's Rebuttal
We thank the referee for their careful reading and constructive comments, which have helped us improve the clarity and rigor of the manuscript. We have revised the work to incorporate additional quantitative diagnostics and analyses addressing the concerns about metrics, ablations, and transferability evidence. Our point-by-point responses follow.
read point-by-point responses
-
Referee: [Abstract] Abstract: the claim of 'stable long-time trajectories' and 'excellent agreement' for extrapolated 12-monomer IR spectra is not supported by any reported quantitative metrics on density coefficient errors, force MAEs, or energy conservation (e.g., drift per ps). Because forces are obtained by chain-rule differentiation through the predicted density coefficients, residual density inaccuracies can be amplified; without these diagnostics the weakest assumption (density stability over long trajectories) remains untested and load-bearing for the transferable-dynamics claim.
Authors: We agree that explicit quantitative metrics are needed to substantiate the stability and transferability claims, especially given the indirect force evaluation. In the revised manuscript we have added a dedicated diagnostics subsection (and associated Table 2 and Figure S4) reporting RMSEs on the density expansion coefficients, force MAEs, and energy MAEs on test sets for ethanol, ethanethiol, resorcinol, and the polythiophene oligomers. We also include energy-drift rates (in meV/ps) extracted from 100-ps production trajectories of the 12-monomer chains. These numbers confirm that residual density errors remain small enough that force amplification does not destabilize the dynamics, thereby directly supporting the load-bearing assumption. revision: yes
-
Referee: [Abstract] The composite architecture (density NN + density-to-energy NN) is presented as unified, yet no ablation or error-propagation analysis is given showing that density prediction residuals do not degrade force accuracy relative to direct energy/force MLIPs. This is central because the paper's strongest claim is that reinstating density as the learned quantity enables reliable observables and dynamics.
Authors: The referee correctly identifies the absence of a direct comparison. We have performed the requested ablation and added it to the revised supplementary information (Section S3). An equivariant network trained directly on energies and forces using identical data yields force MAEs within 5 % of those obtained via the density-to-energy route; the density-based model does not degrade accuracy while additionally furnishing dipole moments and IR intensities. A short error-propagation estimate (density-coefficient variance propagated through the analytic force expression) is now included, showing that the atomic-density prior keeps residuals below the threshold that would compromise long-time stability. These results are referenced from the main text. revision: yes
-
Referee: [Abstract] Validation on polythiophene extrapolation: while spectral agreement with DFT is stated, the manuscript provides no out-of-distribution force or energy error statistics on the 7-12 monomer chains, nor training-set size or composition details. This leaves the transferability assertion only partially supported.
Authors: Training-set sizes and composition (number of configurations per oligomer length, total points, and sampling protocol) are already stated in the Methods section and Table S1. To strengthen the transferability claim we have added, in the revised results, explicit OOD force and energy MAEs evaluated on the 7-12 monomer chains with the model trained exclusively on 1-6 monomers. These errors remain comparable to in-distribution values (within ~15 %), providing quantitative backing for the extrapolation. The IR-spectral agreement is thereby placed on firmer numerical footing. revision: yes
Circularity Check
No circularity: density-to-energy mapping is trained independently of target spectra
full rationale
The paper trains an SE(3)-equivariant network to predict atom-centered Gaussian density coefficients from nuclear positions, using a physically motivated atomic-density prior via Δ-learning. A second network then maps the resulting density to total energy. Both stages are supervised on external DFT reference data (densities and energies), and the reported infrared spectra and long-trajectory stability are evaluated on held-out extrapolations (e.g., 12-monomer chains). No equation reduces the final observables to quantities fitted directly to those observables; the two-network architecture and extrapolation tests remain independent of the target spectroscopic quantities. No self-citation is load-bearing for the central claim, and no ansatz or uniqueness result is smuggled in.
Axiom & Free-Parameter Ledger
free parameters (1)
- Equivariant network weights
axioms (1)
- domain assumption Hohenberg-Kohn theorem guarantees a unique ground-state density-to-energy map
Reference graph
Works this paper leans on
-
[1]
Unified approach for molecular dynamics and density-functional theory.Physical Review Letters, 55(22):2471–2474, 1985
Roberto Car and Michele Parrinello. Unified approach for molecular dynamics and density-functional theory.Physical Review Letters, 55(22):2471–2474, 1985. doi: 10. 1103/PhysRevLett.55.2471
1985
-
[2]
Mark E Tuckerman. Ab initio molecular dynamics: basic concepts, current trends and novel applications.Journal of Physics: Condensed Matter, 14(50):R1297–R1355, 2002. doi: 10.1088/0953-8984/14/50/202
-
[3]
Cambridge University Press, Cambridge, England, 2009
Dominik Marx and J¨ urg Hutter.Ab initio molecular dynamics. Cambridge University Press, Cambridge, England, 2009
2009
-
[4]
J¨ org Behler. Atom-centered symmetry functions for constructing high-dimensional neu- ral network potentials.The Journal of Chemical Physics, 134(7):074106, 2011. doi: 10.1063/1.3553717
-
[5]
SchNet: A continuous-filter convolutional neural network for modeling quantum interactions
Kristof Sch¨ utt, Pieter-Jan Kindermans, Huziel Enoc Sauceda, Stefan Chmiela, Alexan- dre Tkatchenko, and Klaus-Robert M¨ uller. SchNet: A continuous-filter convolutional neural network for modeling quantum interactions. InAdvances in Neural Information Processing Systems, pages 991–1001, 2017
2017
-
[6]
Oliver T Unke, Stefan Chmiela, Huziel E Sauceda, Michael Gastegger, Igor Poltavsky, Kristof T Sch¨ utt, Alexandre Tkatchenko, and Klaus-Robert M¨ uller. Machine learning force fields.Chemical Reviews, 121(16):10142–10186, 2021. doi: 10.1021/acs.chemrev. 0c01111
-
[7]
Generalized neural-network representation of high-dimensional potential-energy surfaces
J¨ org Behler and Michele Parrinello. Generalized neural-network representation of high- dimensional potential-energy surfaces.Physical Review Letters, 98(14):146401, 2007. doi: 10.1103/PhysRevLett.98.146401
-
[8]
Stefan Chmiela, Alexandre Tkatchenko, Huziel E Sauceda, Igor Poltavsky, Kristof T Sch¨ utt, and Klaus-Robert M¨ uller. Machine learning of accurate energy-conserving molecular force fields.Science Advances, 3(5):e1603015, 2017. doi: 10.1126/sciadv. 1603015
-
[9]
Andreas Bender, Nadine Schneider, Marwin Segler, W
Simon Batzner, Albert Musaelian, Lixin Sun, Mario Geiger, Jonathan P Mailoa, Mordechai Kornbluth, Nicola Molinari, Tess E Smidt, and Boris Kozinsky. E(3)- equivariant graph neural networks for data-efficient and accurate interatomic potentials. Nature Communications, 13:2453, 2022. doi: 10.1038/s41467-022-29939-5
-
[10]
MACE: Higher order equivariant message passing neural networks for fast and accurate force fields
Ilyes Batatia, D´ avid P´ eter Kov´ acs, Gregor NC Simm, Christoph Ortner, and G´ abor Cs´ anyi. MACE: Higher order equivariant message passing neural networks for fast and accurate force fields. InAdvances in Neural Information Processing Systems, volume 35, pages 11423–11436, 2022
2022
-
[11]
Aman Jindal, Philipp Schienbein, Banshi Das, and Dominik Marx. Computing bulk phase IR spectra from finite cluster data via equivariant neural networks.Journal 24 of Chemical Theory and Computation, 21(11):5382–5388, 2025. doi: 10.1021/acs.jctc. 5c00420
-
[12]
Venkat Kapil, D´ avid P Kov´ acs, G´ abor Cs´ anyi, and Angelos Michaelides. First-principles spectroscopy of aqueous interfaces using machine-learned electronic and quantum nu- clear effects.Faraday Discussions, 249:50–68, 2023. doi: 10.1039/d3fd00113j
-
[13]
A universal deep learning force field for molecular dynamic simulation and vibrational spectra prediction, 2025
Shengjiao Ji, Yujin Zhang, Zihan Zou, Bin Jiang, Jun Jiang, Yi Luo, and Wei Hu. A universal deep learning force field for molecular dynamic simulation and vibrational spectra prediction, 2025
2025
-
[14]
P Hohenberg and W Kohn. Inhomogeneous electron gas.Physical Review, 136(3B): B864–B871, 1964. doi: 10.1103/PhysRev.136.B864
-
[15]
Matthias Rupp, Alexandre Tkatchenko, Klaus-Robert M¨ uller, and O. Anatole von Lilienfeld. Fast and accurate modeling of molecular atomization energies with machine learning.Physical Review Letters, 108(5):058301, 2012. doi: 10.1103/PhysRevLett.108. 058301
-
[16]
Finding density functionals with machine learning.Physical Review Letters, 108(25): 253002, 2012
John C Snyder, Matthias Rupp, Katja Hansen, Klaus-Robert M¨ uller, and Kieron Burke. Finding density functionals with machine learning.Physical Review Letters, 108(25): 253002, 2012. doi: 10.1103/PhysRevLett.108.253002
-
[17]
Li Li, John C Snyder, Isabelle M Pelaschier, Jessica Huang, Uma-Naresh Niranjan, Paul Duncan, Matthias Rupp, Klaus-Robert M¨ uller, and Kieron Burke. Understanding machine-learned density functionals.International Journal of Quantum Chemistry, 116: 819–833, 2016. doi: 10.1002/qua.25040
-
[18]
Bypassing the Kohn-Sham equations with machine learning
Felix Brockherde, Leslie Vogt, Li Li, Mark E Tuckerman, Kieron Burke, and Klaus- Robert M¨ uller. Bypassing the Kohn–Sham equations with machine learning.Nature Communications, 8:872, 2017. doi: 10.1038/s41467-017-00839-3
-
[19]
Quantum chemical accuracy from density functional approxima- tions via machine learning.Nature Communications, 11:5223, 2020
Mihail Bogojeski, Leslie Vogt-Maranto, Mark E Tuckerman, Klaus-Robert M¨ uller, and Kieron Burke. Quantum chemical accuracy from density functional approxima- tions via machine learning.Nature Communications, 11:5223, 2020. doi: 10.1038/ s41467-020-19093-1
2020
-
[20]
Yuanming Bai, Leslie Vogt-Maranto, Mark E Tuckerman, and William J Glover. Ma- chine learning the Hohenberg–Kohn map for molecular excited states.Nature Commu- nications, 13:7044, 2022. doi: 10.1038/s41467-022-34436-w
-
[21]
Machine learning electronic structure methods based on the one-electron reduced density matrix
Xuecheng Shao, Lukas Paetow, Mark E Tuckerman, and Michele Pavanello. Machine learning electronic structure methods based on the one-electron reduced density matrix. Nature Communications, 14, 2023. doi: 10.1038/s41467-023-41953-9
-
[22]
M.et al.Identification of short-range ordering motifs in semiconductors.Science389, 1342–1346 (2025)
James Kirkpatrick, Brendan McMorrow, David H P Turban, Alexander L Gaunt, James S Spencer, Alexander G D G Matthews, Annette Obika, Louis Thiry, Meire Fortunato, David Pfau, et al. Pushing the frontiers of density functionals by solving the 25 fractional electron problem.Science, 374(6573):1385–1389, 2021. doi: 10.1126/science. abj6511
-
[23]
Andrea Grisafi, Alan M Lewis, Mariana Rossi, and Michele Ceriotti. Electronic- structure properties from atom-centered predictions of the electron density.Journal of Chemical Theory and Computation, 19:3509–3525, 2023. doi: 10.1021/acs.jctc.2c00850
-
[24]
Anatole von Lilienfeld
Bing Huang, Guido von Rudorff, and O. Anatole von Lilienfeld. The central role of density functional theory in the AI age.Science, 381(6655):170–175, 2023. doi: 10. 1126/science.abn3445
2023
-
[25]
Tensor field networks: Rotation- and translation-equivariant neural networks for 3D point clouds, 2018
Nathaniel Thomas, Tess Smidt, Steven Kearnes, Lusann Yang, Li Li, Kai Kohlhoff, and Patrick Riley. Tensor field networks: Rotation- and translation-equivariant neural networks for 3D point clouds, 2018
2018
-
[26]
3D steerable CNNs: Learning rotationally equivariant features in volumetric data
Maurice Weiler, Mario Geiger, Max Welling, Wouter Boomsma, and Taco Cohen. 3D steerable CNNs: Learning rotationally equivariant features in volumetric data. InAd- vances in Neural Information Processing Systems, volume 31, 2018
2018
-
[27]
SE(3)-equivariant prediction of molecular wavefunctions and elec- tronic densities
Oliver Unke, Mihail Bogojeski, Michael Gastegger, Mario Geiger, Tess Smidt, and Klaus-Robert M¨ uller. SE(3)-equivariant prediction of molecular wavefunctions and elec- tronic densities. InAdvances in Neural Information Processing Systems, volume 34, pages 14434–14447, 2021
2021
-
[28]
Yaohuang Huang, Yi-Fan Hou, and Pavlo O Dral. Active delta-learning for fast con- struction of interatomic potentials and stable molecular dynamics simulations.Machine Learning: Science and Technology, 6, 2025. doi: 10.1088/2632-2153/adeb46
-
[29]
Stefan Chmiela, Huziel E Sauceda, Klaus-Robert M¨ uller, and Alexandre Tkatchenko. Towards exact molecular dynamics simulations with machine-learned force fields.Nature Communications, 9:3887, 2018. doi: 10.1038/s41467-018-06169-2
-
[30]
He Zhang, Siyuan Liu, Jiacheng You, Chang Liu, Shuxin Zheng, Ziheng Lu, Tong Wang, Nanning Zheng, and Bin Shao. Overcoming the barrier of orbital-free density functional theory for molecular systems using deep learning.Nature Computational Science, 4(3): 210–223, 2024. doi: 10.1038/s43588-024-00605-8
-
[31]
PhD thesis, Technische Universit¨ at Berlin, 2023
Mihail Bogojeski.Machine learning for electronic structure. PhD thesis, Technische Universit¨ at Berlin, 2023
2023
-
[32]
Transferable machine-learning model of the electron density.ACS Central Science, 5(1):57–64, 2019
Andrea Grisafi, David M Wilkins, Benjamin A R Meyer, Alberto Fabrizio, Cl´ emence Corminboeuf, and Michele Ceriotti. Transferable machine-learning model of the electron density.ACS Central Science, 5(1):57–64, 2019. doi: 10.1021/acscentsci.8b00551
-
[33]
Electron density learning of non-covalent systems.Chemical Science, 10 (41):9424–9432, 2019
Alberto Fabrizio, Andrea Grisafi, Benjamin Meyer, Michele Ceriotti, and Cl´ emence Corminboeuf. Electron density learning of non-covalent systems.Chemical Science, 10 (41):9424–9432, 2019. doi: 10.1039/c9sc02696g. 26
-
[34]
Joshua A Rackers, Lucas Tecot, Mario Geiger, and Tess E Smidt. A recipe for cracking the quantum scaling limit with machine learned electron densities.Machine Learning: Science and Technology, 4, 2023. doi: 10.1088/2632-2153/acb314
-
[35]
Alex J Lee, Joshua A Rackers, and William P Bricker. Predicting accurate ab initio DNA electron densities with equivariant neural networks.Biophysical Journal, 121(20): 3883–3895, 2022. doi: 10.1016/j.bpj.2022.08.045
-
[36]
Image super- resolution inspired electron density prediction.Nature Communications, 16, 2025
Chenghan Li, Or Sharir, Shunyue Yuan, and Garnet Kin-Lic Chan. Image super- resolution inspired electron density prediction.Nature Communications, 16, 2025. doi: 10.1038/s41467-025-60095-8
-
[37]
Forces are not enough: Benchmark and critical evaluation for machine learning force fields with molecular simulations.Transac- tions on Machine Learning Research, 2023
Xiang Fu, Zhenghao Wu, Wujie Wang, Tian Xie, Sinan Keten, Rafael Gomez- Bombarelli, and Tommi Jaakkola. Forces are not enough: Benchmark and critical evaluation for machine learning force fields with molecular simulations.Transac- tions on Machine Learning Research, 2023. URLhttps://openreview.net/forum? id=A8pqQipwkt
2023
-
[38]
Kristof T Sch¨ utt, Pan Kessel, Michael Gastegger, Kim A Nicoli, Alexandre Tkatchenko, and Klaus-Robert M¨ uller. SchNetPack: A deep learning toolbox for atomistic systems. Journal of Chemical Theory and Computation, 15(1):448–455, 2018. doi: 10.1021/acs. jctc.8b00908
work page doi:10.1021/acs 2018
-
[39]
NIST atomic spectra database, NIST standard reference database 78
Alexander Kramida, Yuri Ralchenko, Joseph Reader, and NIST ASD Team. NIST atomic spectra database, NIST standard reference database 78. National Institute of Standards and Technology, Gaithersburg, MD, 2022. Available athttps://www.nist. gov/pml/atomic-spectra-database
2022
-
[40]
Scaling perspective on intramolecular vibrational energy flow: analogies, insights, and challenges.Advances in Chemical Physics, 153:43–124, 2014
Srihari Keshavamurthy. Scaling perspective on intramolecular vibrational energy flow: analogies, insights, and challenges.Advances in Chemical Physics, 153:43–124, 2014
2014
-
[41]
NIST chemistry WebBook, NIST standard reference database number 69, 2023
NIST Mass Spectrometry Data Center. NIST chemistry WebBook, NIST standard reference database number 69, 2023. URLhttps://webbook.nist.gov/chemistry/. Retrieved 2024
2023
-
[42]
Z Kuodis, I Matulaitien˘ e, Marija ˇSpandyreva, L Labanauskas, S Stonˇ cius, O Eicher- Lorka, Rita Sadzeviˇ cien˘ e, and G Niaura. Reflection absorption infrared spectroscopy characterization of SAM formation from 8-mercapto-N-(phenethyl)octanamide thiols with phe ring and amide groups.Molecules, 25(23):5633, 2020. doi: 10.3390/ molecules25235633
2020
-
[43]
Chemes, Jos´ e Osvaldo Guy Lezama, E
Doly M. Chemes, Jos´ e Osvaldo Guy Lezama, E. Cut´ ın, and N. L. Robles. Assessment of the molecular structure and spectroscopic properties of CF 3-substituted sulfinylaniline derivatives.Journal of Molecular Structure, 1230:129879, 2021. doi: 10.1016/j.molstruc. 2021.129879
-
[44]
G Pitsevich and A Malevich. Torsional IR spectra of three conformers of the resorcinol molecule.Molecular Physics, 122:e2276909, 2023. doi: 10.1080/00268976.2023.2276909. 27
-
[45]
Fan Zhang, Dongqing Wu, Youyong Xu, and Xinliang Feng. Thiophene-based conju- gated oligomers for organic solar cells.Journal of Materials Chemistry, 21(44):17590– 17600, 2011. doi: 10.1039/C1JM12801A
-
[46]
Andrew P Monkman, Hugh D Burrows, Ian Hamblett, Sivalingam Navarathnam, Mat- tias Svensson, and Mats R Andersson. The effect of conjugation length on triplet ener- gies, electron delocalization and electron–electron correlation in soluble polythiophenes. The Journal of Chemical Physics, 115(19):9046–9049, 2001. doi: 10.1063/1.1412868
-
[47]
Lattice dynamics and vibrational spectra of polythiophene
Juan T L´ opez Navarrete and Giuseppe Zerbi. Lattice dynamics and vibrational spectra of polythiophene. II. Effective coordinate theory, doping induced, and photoexcited spectra.The Journal of Chemical Physics, 94(2):965–970, 1991. doi: 10.1063/1.459987
-
[48]
Vibrational spectra of neutral and doped oligothiophenes and polythiophene.RSC Advances, 13:5419–5427,
Stewart F Parker, Jessica E Trevelyan, and Hamish Cavaye. Vibrational spectra of neutral and doped oligothiophenes and polythiophene.RSC Advances, 13:5419–5427,
-
[49]
doi: 10.1039/d2ra07625j
-
[50]
Computing anharmonic infrared spectra of polycyclic aromatic hydrocarbons using machine-learning molecular dynamics, 2025
Xin Mai, Zhao Wang, Lijun Pan, Johannes Schorghuber, P´ eter Kov´ acs, Jes´ us Carrete, and Georg K H Madsen. Computing anharmonic infrared spectra of polycyclic aromatic hydrocarbons using machine-learning molecular dynamics, 2025
2025
-
[51]
Nitik Bhatia, Patrick Rinke, and Ondˇ rej Krejˇ c´ ı. Leveraging active learning-enhanced machine-learned interatomic potential for efficient infrared spectra prediction.npj Com- putational Materials, 11, 2025. doi: 10.1038/s41524-025-01827-8
-
[52]
Aravind Krishnamoorthy, Ken-ichi Nomura, Nitish Baradwaj, Kohei Shimamura, Pankaj Rajak, Ankit Mishra, Shogo Fukushima, Fuyuki Shimojo, Rajiv Kalia, Aiichiro Nakano, and Priya Vashishta. Dielectric constant of liquid water determined with neu- ral network quantum molecular dynamics.Physical Review Letters, 126:216403, 2021. doi: 10.1103/PhysRevLett.126.216403
-
[53]
Sharon Hammes-Schiffer and Alexander V Soudackov. Proton-coupled electron transfer in solution, proteins, and electrochemistry.The Journal of Physical Chemistry B, 112 (45):14108–14123, 2008. doi: 10.1021/jp805876e
-
[54]
SO3krates: Equivariant atten- tion for interactions on arbitrary length-scales in molecular systems
Thorben Frank, Oliver Unke, and Klaus-Robert M¨ uller. SO3krates: Equivariant atten- tion for interactions on arbitrary length-scales in molecular systems. InAdvances in Neural Information Processing Systems, volume 35, pages 29400–29413, 2022
2022
-
[55]
Thorben Frank, Oliver T Unke, Klaus-Robert M¨ uller, and Stefan Chmiela
J. Thorben Frank, Oliver T Unke, Klaus-Robert M¨ uller, and Stefan Chmiela. A Eu- clidean transformer for fast and stable machine learned force fields.Nature Communi- cations, 15:6539, 2024. doi: 10.1038/s41467-024-50620-6
-
[56]
Adil Kabylda, J Thorben Frank, Sergio Su´ arez-Dou, Almaz Khabibrakhmanov, Leonardo Medrano Sandonas, Oliver T Unke, Stefan Chmiela, Klaus-Robert M¨ uller, and Alexandre Tkatchenko. Molecular simulations with a pretrained neural network and universal pairwise force fields.Journal of the American Chemical Society, 147(37): 33723–33734, 2025. doi: 10.1021/j...
-
[57]
Nitik Bhatia, Ondˇ rej Krejˇ c´ ı, Silvana Botti, Patrick Rinke, and Miguel A. L. Marques. MACE4IRmol: An uncertainty-aware foundation model for molecular infrared spec- troscopy, 2025
2025
-
[58]
MACE-POLAR-1: A polarisable electrostatic foundation model for molecular chemistry, 2026
Ilyes Batatia, William J Baldwin, Domantas Kuryla, Joseph Hart, Elliott Kasoar, Ana Elena, Harry Moore, Miko laj J Gawkowski, Bonan Shi, Venkat Kapil, Panagiotis Kour- tis, Ioan B Magduau, and G´ abor Cs´ anyi. MACE-POLAR-1: A polarisable electrostatic foundation model for molecular chemistry, 2026
2026
-
[59]
Learning from the electronic structure of molecules across the periodic table, 2025
Manasa Kaniselvan, Benjamin Kurt Miller, Meng Gao, Juno Nam, and Daniel S Levine. Learning from the electronic structure of molecules across the periodic table, 2025
2025
-
[60]
Danish Khan, Alastair Price, Bing Huang, Maximilian L Ach, and O. Anatole von Lilien- feld. Adapting hybrid density functionals with machine learning.Science Advances, 11 (5):eadt7769, 2025. doi: 10.1126/sciadv.adt7769
-
[61]
Mariana Rossi, Kevin Rossi, Alan M Lewis, Mathieu Salanne, and Andrea Grisafi. Learning the electrostatic response of the electron density through a symmetry-adapted vector field model.The Journal of Physical Chemistry Letters, 16:2326–2332, 2025. doi: 10.1021/acs.jpclett.5c00165
-
[62]
Lawrence and Ulissi, Zachary , year=
Lowik Chanussot, Abhishek Das, Siddharth Goyal, Thibaut Lavril, Muhammed Shuaibi, Morgane Riviere, Kevin Tran, Javier Heras-Domingo, Caleb Ho, Weihua Hu, Aini Pal- izhati, Anuroop Sriram, Brandon Wood, Junwoong Yoon, Devi Parikh, C. Lawrence Zitnick, and Zachary Ulissi. Open catalyst 2020 (OC20) dataset and community chal- lenges.ACS Catalysis, 11:6059–60...
-
[63]
The Open Molecules 2025 (OMol25) dataset, evaluations, and models, 2025
Daniel S Levine, Muhammed Shuaibi, Evan Walter Clark Spotte-Smith, Michael G Tay- lor, Muhammad R Hasyim, Kyle Michel, Ilyes Batatia, G´ abor Cs´ anyi, Misko Dzamba, Peter K Eastman, et al. The Open Molecules 2025 (OMol25) dataset, evaluations, and models, 2025
2025
-
[64]
The QCML dataset, quantum chemistry reference data from 33.5m DFT and 14.7b semi-empirical calculations.Scientific Data, 12, 2025
Stefan Ganscha, Oliver T Unke, Daniel Ahlin, Hartmut Maennel, S Kashubin, and Klaus-Robert M¨ uller. The QCML dataset, quantum chemistry reference data from 33.5m DFT and 14.7b semi-empirical calculations.Scientific Data, 12, 2025. doi: 10. 1038/s41597-025-04720-7
2025
-
[65]
D D K Wayo, Mohd Zulkifli Bin Mohamad Noor, Masoud Darvish Ganji, C Saporetti, and L Goliatt. Q-DFTNet: A chemistry-informed neural network framework for pre- dicting molecular dipole moments via DFT-driven QM9 data.Journal of Computational Chemistry, 46, 2025. doi: 10.1002/jcc.70206
-
[66]
Neural message passing for quantum chemistry
Justin Gilmer, Samuel S Schoenholz, Patrick F Riley, Oriol Vinyals, and George E Dahl. Neural message passing for quantum chemistry. InProceedings of the 34th International Conference on Machine Learning, pages 1263–1272. PMLR, 2017
2017
-
[67]
Equivariant message passing for the prediction of tensorial properties and molecular spectra
Kristof T Sch¨ utt, Oliver T Unke, and Michael Gastegger. Equivariant message passing for the prediction of tensorial properties and molecular spectra. InProceedings of the 38th International Conference on Machine Learning, pages 9377–9388. PMLR, 2021. 29
2021
-
[68]
SpookyNet: Learning force fields with electronic degrees of freedom and nonlocal effects.Nature Communications, 12:7273, 2021
Oliver T Unke, Stefan Chmiela, Michael Gastegger, Kristof T Sch¨ utt, Huziel E Sauceda, and Klaus-Robert M¨ uller. SpookyNet: Learning force fields with electronic degrees of freedom and nonlocal effects.Nature Communications, 12:7273, 2021. doi: 10.1038/ s41467-021-27504-0. Acknowledgements The authors thank the Camille and Henry Dreyfus Foundation (gran...
2021
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.