Recognition: unknown
Learning Structure, Energy, and Dynamics: A Survey of Artificial Intelligence for Protein Dynamics
Pith reviewed 2026-05-07 14:08 UTC · model grok-4.3
The pith
Artificial intelligence for protein dynamics organizes into three perspectives: learning structures and trajectories, incorporating energy signals, and accelerating simulations.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
The paper states that advances in AI for protein dynamics fall into three perspectives—learning from structural ensembles and trajectories, learning from physical energy signals, and learning to accelerate molecular simulations—and reviews representative techniques including conformation ensemble generation, trajectory generation, Boltzmann generators, physics-aware adaptation, machine learning potentials, coarse-grained modeling, and collective variable discovery, while discussing datasets and open challenges in scalability, thermodynamic consistency, kinetic fidelity, and experimental integration.
What carries the argument
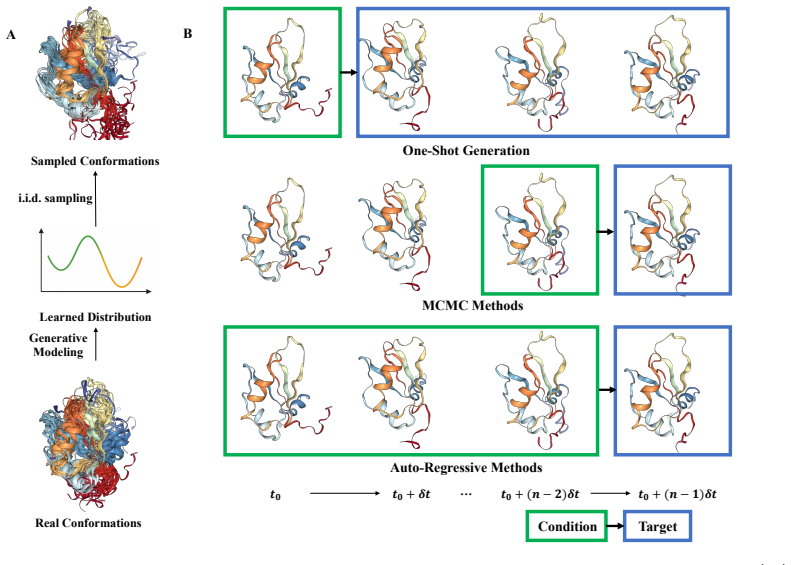
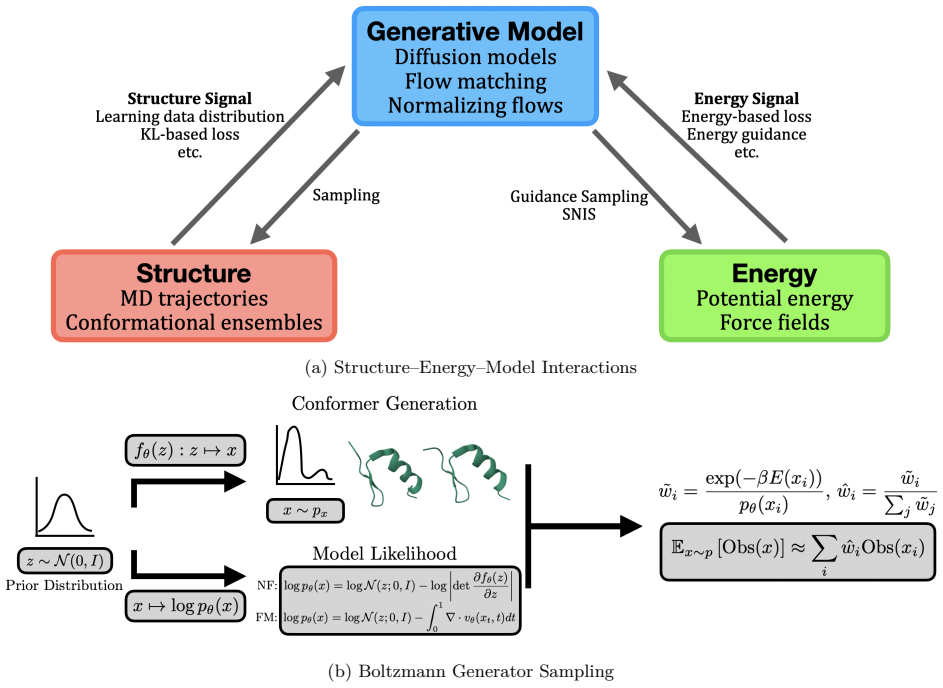
The three-perspective classification that sorts AI methods according to their primary focus on structural data, energy landscapes, or simulation speedup.
If this is right
- Better generation of structural ensembles and trajectories would let researchers identify functional protein states that are hard to observe directly.
- Energy-aware methods would produce predictions that automatically follow physical laws and reduce unphysical artifacts.
- AI-driven acceleration of simulations would extend the reachable time scales to those relevant for many biological processes.
- Resolving the listed challenges would make it easier to combine these computational tools with actual experimental measurements.
Where Pith is reading between the lines
- This grouping could help practitioners pick the most suitable AI approach for a given protein system or question.
- Future models might blend all three perspectives into single frameworks that handle structure, energy, and speed at once.
- The highlighted challenges could serve as a short list of priorities for new method development.
Load-bearing premise
The chosen examples and challenges together give a complete view of current AI work on protein dynamics with no important omissions.
What would settle it
Locating a widely used AI method for protein movements that fits none of the three perspectives or uncovering a major practical barrier the survey does not mention would show the overview is incomplete.
Figures



read the original abstract
Protein dynamics underlie many biological functions, yet remain difficult to characterize due to the high computational cost of molecular dynamics simulations and the scarcity of dynamic structural data. This survey reviews recent advances in artificial intelligence for protein dynamics from three perspectives: learning from structural ensembles and trajectories, learning from physical energy signals, and learning to accelerate molecular simulations. We summarize representative methods for conformation ensemble generation, trajectory generation, Boltzmann generators, physics-aware adaptation, machine learning potentials, coarse-grained modeling, and collective variable discovery. We further discuss available datasets and key open challenges, such as scalability, thermodynamic consistency, kinetic fidelity, and integration with experimental constraints.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript is a survey of artificial intelligence methods for modeling protein dynamics. It organizes recent advances into three perspectives: learning from structural ensembles and trajectories (covering conformation ensemble generation and trajectory generation), learning from physical energy signals (covering Boltzmann generators, physics-aware adaptation, and machine learning potentials), and learning to accelerate molecular simulations (covering coarse-grained modeling and collective variable discovery). The paper also reviews available datasets and identifies open challenges including scalability, thermodynamic consistency, kinetic fidelity, and integration with experimental constraints.
Significance. If the coverage is accurate and reasonably complete, the survey would provide a useful organizational framework for a rapidly evolving interdisciplinary area at the intersection of AI and biophysics. The three-perspective structure helps clarify how data-driven methods can complement or replace traditional molecular dynamics, and the explicit listing of challenges (thermodynamic consistency, kinetic fidelity) could usefully direct future work. The paper does not introduce new methods or proofs, so its value lies in synthesis rather than novel claims.
minor comments (3)
- The abstract states that the survey summarizes 'representative methods' for seven categories, but the main text should include a brief table or explicit list (perhaps in §2 or §4) mapping each cited paper to its primary perspective and method category to improve navigability for readers.
- In the discussion of open challenges, the distinction between thermodynamic consistency and kinetic fidelity is conceptually important but would benefit from one or two concrete examples of how a method can satisfy one while failing the other (e.g., a Boltzmann generator that matches equilibrium distributions but not transition rates).
- The paper mentions 'available datasets' but does not appear to provide a consolidated table of commonly used benchmarks (e.g., specific protein systems, trajectory lengths, or experimental references); adding such a table in the datasets section would strengthen the survey's utility.
Simulated Author's Rebuttal
We thank the referee for their positive assessment of our survey and for recommending minor revision. The referee's summary accurately captures the manuscript's organization into three perspectives on AI for protein dynamics, the covered methods, datasets, and open challenges. Since no specific major comments were provided in the report, we have no point-by-point responses to offer at this stage.
Circularity Check
No circularity: pure survey with no derivations or predictions
full rationale
This is a review paper that organizes and summarizes existing literature on AI methods for protein dynamics across three perspectives (structural ensembles/trajectories, energy signals, simulation acceleration). It presents no original equations, fitted parameters, predictions, or theoretical derivations. All content is descriptive citation of prior work, with no self-referential steps that reduce claims to inputs by construction. The central claim is organizational rather than falsifiable or load-bearing, so no circularity analysis applies.
Axiom & Free-Parameter Ledger
Reference graph
Works this paper leans on
-
[1]
AlphaFold meets flow matching for generating protein ensembles
Bowen Jing, Bonnie Berger, and Tommi Jaakkola. AlphaFold meets flow matching for generating protein ensembles. In Ruslan Salakhutdinov, Zico Kolter, Katherine Heller, Adrian Weller, Nuria Oliver, Jonathan Scarlett, and Felix Berkenkamp, editors,Proceedings of the 41st International Conference on Machine Learning, volume 235 ofProceedings of Machine Learni...
-
[2]
Tan, Joey Bose, Chen Lin, Leon Klein, Michael M
Charlie B. Tan, Joey Bose, Chen Lin, Leon Klein, Michael M. Bronstein, and Alexander Tong. Scalable equilibrium sampling with sequential boltzmann generators. InForty-second International Conference on Machine Learning, 2025a. URLhttps://openreview.net/forum?id=U7eMoRDIGihttps://openreview.net/forum?id=U7eMoRDIGi. Charlie B. Tan, Majdi Hassan, Leon Klein,...
-
[3]
doi: 10.64898/2026.02.10.70487310.64898/2026.02.10.704873. URL https://www.biorxiv.org/content/early/2026/02/11/2026.02.10.704873https://www.biorxiv.org/content/early/2026/02/11/2026.02.10.704873. Tong Wang, Xinheng He, Mingyu Li, Yatao Li, Ran Bi, Yusong Wang, Chaoran Cheng, Xiangzhen Shen, Jiawei Meng, He Zhang, Haiguang Liu, Zun Wang, Shaoning Li, Bin ...
work page doi:10.64898/2026.02.10.70487310.64898/2026.02.10.704873 2026
-
[4]
Ferguson
Wei Chen and Andrew L. Ferguson. Molecular enhanced sampling with autoencoders: On-the-fly collective variable discovery and accelerated free energy landscape exploration.Journal of Computational Chemistry, 39(25):2079–2102, 9
2079
-
[5]
Exploring the conformational ensembles of protein–protein complex with transformer-based generative model.Journal of Chemical Theory and Computation, 20(11):4469– 4480, 2024c
Jianmin Wang, Xun Wang, Yanyi Chu, Chunyan Li, Xue Li, Xiangyu Meng, Yitian Fang, Kyoung Tai No, Jiashun Mao, and Xiangxiang Zeng. Exploring the conformational ensembles of protein–protein complex with transformer-based generative model.Journal of Chemical Theory and Computation, 20(11):4469– 4480, 2024c. Yusong Wang, Youjun Xu, Wentao Li, Haoyu Yu, Wenju...
2026
-
[6]
URLhttps://arxiv.org/abs/2603.17633https://arxiv.org/abs/2603.17633. Sarah Lewis, Tim Hempel, José Jiménez-Luna, Michael Gastegger, Yu Xie, Andrew YK Foong, Victor García Satorras, Osama Abdin, Bastiaan S Veeling, Iryna Zaporozhets, et al. Scalable emulation of protein equilibrium ensembles with generative deep learning.Science, 389(6761):eadv9817,
-
[7]
Physically grounded generative modeling of all-atom biomolecular dynamics.bioRxiv, pages 2026–02, 2026b
Bin Feng, Jiying Zhang, Xinni Zhang, Ming Zhang, Patrick Barth, Zijing Liu, and Yu Li. Physically grounded generative modeling of all-atom biomolecular dynamics.bioRxiv, pages 2026–02, 2026b. Saro Passaro, Gabriele Corso, Jeremy Wohlwend, Mateo Reveiz, Stephan Thaler, Vignesh Ram Somnath, Noah Getz, Tally Portnoi, Julien Roy, Hannes Stark, et al. Boltz-2:...
2026
-
[8]
arXiv preprint arXiv:2509.13294 (2025)
Allan dos Santos Costa, Manvitha Ponnapati, Dana Rubin, Tess Smidt, and Joseph Jacobson. Accelerating protein molecular dynamics simulation with deepjump.arXiv preprint arXiv:2509.13294,
-
[9]
Zirui Fan, Junjie Zhu, and Hai-Feng Chen. Dynafold: A latent diffusion based generative framework for protein dynamic trajectory.bioRxiv, 2025a. Wei Lu, Jixian Zhang, Weifeng Huang, Ziqiao Zhang, Xiangyu Jia, Zhenyu Wang, Leilei Shi, Chengtao Li, Peter G Wolynes, and Shuangjia Zheng. Dynamicbind: predicting ligand-specific protein-ligand complex structure...
-
[10]
Shaoning Li, Yusong Wang, Mingyu Li, Jian Zhang, Bin Shao, Nanning Zheng, and Jian Tang. F3low: Frame-to-frame coarse-grained molecular dynamics with se (3) guided flow matching.arXiv preprint arXiv:2405.00751,
-
[11]
Yaowei Jin, Qi Huang, Ziyang Song, Mingyue Zheng, Dan Teng, and Qian Shi
URLhttps://arxiv.org/abs/2508.03709https://arxiv.org/abs/2508.03709. Yaowei Jin, Qi Huang, Ziyang Song, Mingyue Zheng, Dan Teng, and Qian Shi. P2dflow: A protein ensemble generative model with se(3) flow matching.Journal of Chemical Theory and Computation, 21(6):3288– 3296,
-
[12]
Yuyang Wang, Jiarui Lu, Navdeep Jaitly, Josh Susskind, and Miguel Angel Bautista
URL https://openreview.net/forum?id=PtRLxAAKzKhttps://openreview.net/forum?id=PtRLxAAKzK. Yuyang Wang, Jiarui Lu, Navdeep Jaitly, Josh Susskind, and Miguel Angel Bautista. Simplefold: Folding proteins is simpler than you think.arXiv preprint arXiv:2509.18480, 2025a. Nima Shoghi, Yuxuan Liu, Yuning Shen, Rob Brekelmans, Pan Li, and Quanquan Gu. Scalable sp...
-
[13]
Joseph C Kim, David Bloore, Karan Kapoor, Jun Feng, Ming-Hong Hao, and Mengdi Wang
URLhttps://openreview.net/forum?id=XCTVFJwS9LJhttps://openreview.net/forum?id=XCTVFJwS9LJ. Joseph C Kim, David Bloore, Karan Kapoor, Jun Feng, Ming-Hong Hao, and Mengdi Wang. Scalable normalizing flows enable boltzmann generators for macromolecules.arXiv preprint arXiv:2401.04246,
-
[14]
arXiv preprint arXiv:2409.09787 , year=
URLhttps://arxiv.org/abs/2409.09787https://arxiv.org/abs/2409.09787. Tara Akhound-Sadegh, Jungyoon Lee, Joey Bose, Valentin De Bortoli, Arnaud Doucet, Michael M. Bronstein, Dominique Beaini, Siamak Ravanbakhsh, Kirill Neklyudov, and Alexander Tong. Pro- gressive inference-time annealing of diffusion models for sampling from boltzmann densities. In The Thi...
-
[15]
URLhttps://arxiv.org/abs/2509.03726https://arxiv.org/abs/2509.03726. Christopher von Klitzing, Denis Blessing, Henrik Schopmans, Pascal Friederich, and Gerhard Neumann. Learning boltzmann generators via constrained mass transport. InThe Fourteenth International Confer- ence on Learning Representations,
work page internal anchor Pith review Pith/arXiv arXiv
- [16]
-
[17]
Laurent Dinh, Jascha Sohl-Dickstein, and Samy Bengio
URLhttps://arxiv.org/abs/2602.03729https://arxiv.org/abs/2602.03729. Laurent Dinh, Jascha Sohl-Dickstein, and Samy Bengio. Density estimation using real nvp.arXiv preprint arXiv:1605.08803,
-
[18]
Chan, Lester Hedges, Meritxell Malagarriga, Rolf David, Miguel de la Puente, Damien Laage, Iñaki Tuñón, Marc W
Valentin Gradisteanu, Elliot W. Chan, Lester Hedges, Meritxell Malagarriga, Rolf David, Miguel de la Puente, Damien Laage, Iñaki Tuñón, Marc W. van der Kamp, and Kir- ill Zinovjev. Simulating enzyme catalysis with electrostatically embedded machine learn- ing potentials.ChemRxiv, 2025(0707),
2025
-
[19]
doi: 10.26434/chemrxiv-2025-nw9lt10.26434/chemrxiv-2025-nw9lt. URL https://chemrxiv.org/doi/abs/10.26434/chemrxiv-2025-nw9lthttps://chemrxiv.org/doi/abs/10.26434/chemrxiv-2025-nw9lt. Xujian Wang, Haocheng Tang, Xiongwu Wu, Bernard Brooks, Junmei Wang, and Wan- Lu Li. Redefining computational enzymology with multiscale machine learning/molecular mechanics ...
work page doi:10.26434/chemrxiv-2025-nw9lt10.26434/chemrxiv-2025-nw9lt 2025
-
[20]
Yang et al., MatterSim: A Deep Learning Atomistic Model Across Elements, Temperatures and Pressures
ISSN 2052-4463. Han Yang, Chenxi Hu, Yichi Zhou, Xixian Liu, Yu Shi, Jielan Li, Guanzhi Li, Zekun Chen, Shuizhou Chen, Claudio Zeni, et al. Mattersim: A deep learning atomistic model across elements, temperatures and pressures.arXiv preprint arXiv:2405.04967, 2024a. Haokai Hong, Wanyu Lin, Zhang Chusong, and KC Tan. Geometric graph neural diffusion for st...
-
[21]
29 MohammadM.SultanandVijayS.Pande
URLhttps://arxiv.org/abs/2601.22123https://arxiv.org/abs/2601.22123. 29 MohammadM.SultanandVijayS.Pande. Automateddesignofcollectivevariablesusingsupervisedmachine learning.The Journal of Chemical Physics, 149(9), 9
-
[22]
Learning collective variables from time-lagged generation
Seonghyun Park, Kiyoung Seong, Soojung Yang, Rafael Gomez-Bombarelli, and Sungsoo Ahn. Learning collective variables from time-lagged generation. InICML 2025 Generative AI and Biology (GenBio) Workshop,
2025
-
[23]
Adrià Pérez, Pablo Herrera-Nieto, Stefan Doerr, and Gianni De Fabritiis
doi:10.1109/DLS49591.2019.0000710.1109/DLS49591.2019.00007. Adrià Pérez, Pablo Herrera-Nieto, Stefan Doerr, and Gianni De Fabritiis. Adaptivebandit: A multi-armed bandit framework for adaptive sampling in molecular simulations.Journal of Chemical Theory and Com- putation, 16(7):4685–4693,
work page doi:10.1109/dls49591.2019.0000710.1109/dls49591.2019.00007 2019
-
[24]
arXiv preprint arXiv:2507.17700 (2025)
URL https://arxiv.org/abs/2507.17700https://arxiv.org/abs/2507.17700. Haochuan Chen, Benoît Roux, and Christophe Chipot. Discovering reaction pathways, slow variables, and committor probabilities with machine learning.Journal of Chemical Theory and Computation, 19(14): 4414–4426,
-
[25]
doi:10.1038/s43588-025-00828-310.1038/s43588-025-00828-3
ISSN 2662-8457. doi:10.1038/s43588-025-00828-310.1038/s43588-025-00828-3. Bojun Liu, Jordan G Boysen, Ilona Christy Unarta, Xuefeng Du, Yixuan Li, and Xuhui Huang. Exploring transition states of protein conformational changes via out-of-distribution detection in the hyperspherical latent space.Nature communications, 16(1):349, 1
work page doi:10.1038/s43588-025-00828-310.1038/s43588-025-00828-3
-
[26]
doi:10.1038/s41467-024-55228-410.1038/s41467-024-55228-4
ISSN 2041-1723. doi:10.1038/s41467-024-55228-410.1038/s41467-024-55228-4. Stephan Thaler, Zhiyi Wu, William G. Glass, Richard T. Bradshaw, Gail Bartlett, Prudencio Tossou, and Geoffrey P. F. Wood. Boltz-abfe: Free energy perturbation without crystal structures.Journal of Chemical Theory and Computation, 22(4):1823–1833,
work page doi:10.1038/s41467-024-55228-410.1038/s41467-024-55228-4 2041
-
[27]
Sam Giannakoulias, John Ferrie, and Andrew Apicello
doi:10.1021/acs.jctc.5c0145110.1021/acs.jctc.5c01451. Sam Giannakoulias, John Ferrie, and Andrew Apicello. Fepω: The end of parameter tuning.ChemRxiv, 2025(1023),
work page doi:10.1021/acs.jctc.5c0145110.1021/acs.jctc.5c01451 2025
-
[28]
doi:10.26434/chemrxiv-2025-bg1t910.26434/chemrxiv-2025-bg1t9. Donald J. M. van P and Willem Jespers. Integrating machine learning into free energy per- turbation workflows.Journal of Chemical Information and Modeling, 65(19):9856–9864,
work page doi:10.26434/chemrxiv-2025-bg1t910.26434/chemrxiv-2025-bg1t9 2025
-
[29]
doi:10.1021/acs.jcim.5c0144910.1021/acs.jcim.5c01449. 31 Helen M Berman, John Westbrook, Zukang Feng, Gary Gilliland, Talapady N Bhat, Helge Weissig, Ilya N Shindyalov, and Philip E Bourne. The protein data bank.Nucleic acids research, 28(1):235–242,
work page doi:10.1021/acs.jcim.5c0144910.1021/acs.jcim.5c01449
-
[30]
Alphafold protein structure database in 2024: providing structure coverage for over 214 million protein sequences.Nucleic acids research, 52(D1):D368–D375,
Mihaly Varadi, Damian Bertoni, Paulyna Magana, Urmila Paramval, Ivanna Pidruchna, Malarvizhi Rad- hakrishnan, Maxim Tsenkov, Sreenath Nair, Milot Mirdita, Jingi Yeo, et al. Alphafold protein structure database in 2024: providing structure coverage for over 214 million protein sequences.Nucleic acids research, 52(D1):D368–D375,
2024
-
[31]
Metagenomic-scale analysis of the predicted protein structure universe.bioRxiv, pages 2025–04,
Jingi Yeo, Yewon Han, Nicola Bordin, Andy M Lau, Shaun M Kandathil, Hyunbin Kim, Eli Levy Karin, Milot Mirdita, David T Jones, Christine Orengo, et al. Metagenomic-scale analysis of the predicted protein structure universe.bioRxiv, pages 2025–04,
2025
-
[32]
Danny Reidenbach, Zhonglin Cao, Zuobai Zhang, Kieran Didi, Tomas Geffner, Guoqing Zhou, Jian Tang, Christian Dallago, Arash Vahdat, Emine Kucukbenli, et al. Consistent synthetic sequences unlock struc- tural diversity in fully atomistic de novo protein design.arXiv preprint arXiv:2512.01976,
-
[33]
Ce Liu, Jun Wang, Zhiqiang Cai, Yingxu Wang, Huizhen Kuang, Kaihui Cheng, Liwei Zhang, Qingkun Su, Yining Tang, Fenglei Cao, et al. Dynamic pdb: A new dataset and a se (3) model extension by integrating dynamic behaviors and physical properties in protein structures.arXiv preprint arXiv:2408.12413,
-
[34]
Af-calvados: Alphafold-guided simulations of multi-domain proteins at the proteome level.bioRxiv, pages 2025–10,
Sören von Bülow, Kristoffer E Johansson, and Kresten Lindorff-Larsen. Af-calvados: Alphafold-guided simulations of multi-domain proteins at the proteome level.bioRxiv, pages 2025–10,
2025
-
[35]
Enhanced sampling, public dataset and generative model for drug-protein dissociation dynamics
Maodong Li, Jiying Zhang, Bin Feng, Wenqi Zeng, Dechin Chen, Zhijun Pan, Yu Li, Zijing Liu, and Yi Isaac Yang. Enhanced sampling, public dataset and generative model for drug-protein dissociation dynamics. arXiv preprint arXiv:2504.18367,
-
[36]
Mobidb in 2025: inte- grating ensemble properties and function annotations for intrinsically disordered proteins.Nucleic Acids Research, 53(D1):D495–D503,
Damiano Piovesan, Alessio Del Conte, Mahta Mehdiabadi, Maria Cristina Aspromonte, Matthias Blum, Giulio Tesei, Sören von Bülow, Kresten Lindorff-Larsen, and Silvio CE Tosatto. Mobidb in 2025: inte- grating ensemble properties and function annotations for intrinsically disordered proteins.Nucleic Acids Research, 53(D1):D495–D503,
2025
-
[37]
Uniprot: the universal protein knowledgebase in 2025.Nucleic Acids Research, 53(D1):D609–D617, November
Alex Bateman, Maria-Jesus Martin, Sandra Orchard, Michele Magrane, Aduragbemi Adesina, Shadab Ah- mad, Emily H Bowler-Barnett, Hema Bye-A-Jee, David Carpentier, Paul Denny, Jun Fan, Penelope Garmiri, Leonardo Jose da Costa Gonzales, Abdulrahman Hussein, Alexandr Ignatchenko, Giuseppe In- sana, Rizwan Ishtiaq, Vishal Joshi, Dushyanth Jyothi, Swaathi Kandas...
2025
-
[38]
doi:10.1093/nar/gkae101010.1093/nar/gkae1010
ISSN 1362-4962. doi:10.1093/nar/gkae101010.1093/nar/gkae1010. URL http://dx.doi.org/10.1093/nar/gkae1010http://dx.doi.org/10.1093/nar/gkae1010. Rommie E. Amaro, Johan Åqvist, Ivet Bahar, Federica Battistini, Adam Bellaiche, Daniel Beltran, Philip C. Biggin, Massimiliano Bonomi, Gregory R. Bowman, Richard A. Bryce, Giovanni Bussi, Paolo Carloni, DavidA.Cas...
-
[39]
doi: 10.1063/5.003652210.1063/5.0036522
ISSN 1089-7690. doi: 10.1063/5.003652210.1063/5.0036522. URL http://dx.doi.org/10.1063/5.0036522http://dx.doi.org/10.1063/5.0036522. 34
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.