Recognition: unknown
Robust Clustering Analysis of Genes Related to Age-related Macular Degeneration using RNA-Seq
Pith reviewed 2026-05-07 14:01 UTC · model grok-4.3
The pith
A generalized gene co-expression method applied to AMD RNA-Seq data identifies stable modules and previously unknown hub genes.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
By generalizing Multiscale Embedded Gene Co-Expression Network Analysis with curated module quality metrics to select statistical distance or information-theoretic similarity measures, adding a stability test for hub genes under noise, and introducing differential module eigengene analysis, the work detects robust hub genes and modules in AMD RNA-Seq data that align with earlier reports while also revealing previously undiscovered hub genes that may illuminate disease mechanisms and support new treatment development.
What carries the argument
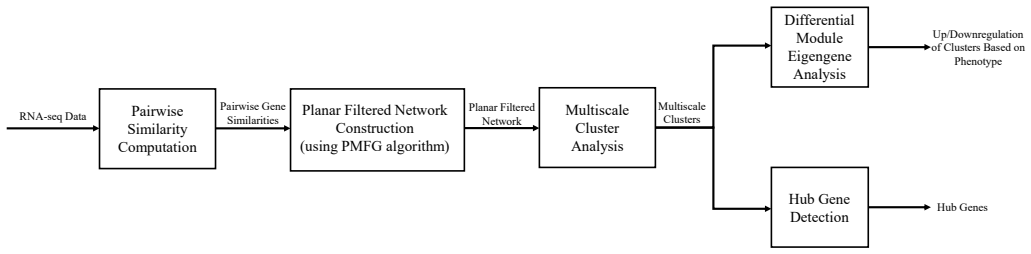
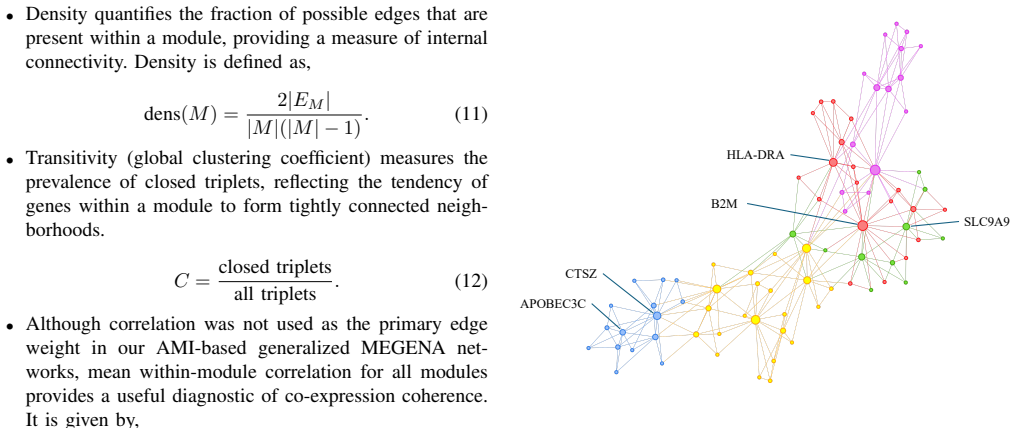
Generalized MEGENA framework that uses module quality evaluation metrics to pick similarity measures, runs a stability test on hub genes, and performs differential eigengene analysis to compare disease versus control modules.
If this is right
- The detected modules provide a validated network view that links many known AMD genes into coordinated groups.
- The newly identified hub genes become concrete candidates for follow-up mechanistic studies and drug-target screening.
- The stability test reduces the chance that reported hubs arise only from sample-specific noise.
- Differential eigengene analysis directly shows which modules are up- or down-regulated in patients relative to controls.
Where Pith is reading between the lines
- The same pipeline could be rerun on larger or multi-omics AMD datasets to test whether the new hubs remain stable.
- If the undiscovered hubs prove functionally relevant in cell or animal models, they may open pathways not currently targeted by existing AMD therapies.
- Extending the stability test to cross-dataset comparisons could further strengthen in the modules before they are used for biomarker development.
Load-bearing premise
The selected similarity measures and stability checks actually reflect true biological gene relationships instead of artifacts created by RNA-Seq technical noise or arbitrary parameter choices.
What would settle it
Independent RNA-Seq or qPCR datasets in which the newly reported hub genes show no consistent differential expression or functional enrichment in AMD tissue, or in which the stability test fails to recover the same modules across repeated random subsamples of the original data.
Figures

read the original abstract
Identifying genes associated with diseases is crucial to understanding disease mechanisms and developing therapies. However, identification of individual genes associated with a disease often needs to be supplemented with clustering analysis to understand the relationships between genes and identify gene modules beyond individual gene-level relationships. Gene co-expression networks are widely used as a graph theoretic approach to the clustering analysis of genes. In our work, we perform robust clustering analysis on RNA-Seq data of Age-related Macular Degeneration (AMD) patients and controls by generalizing one such framework, Multiscale Embedded Gene Co-Expression Network Analysis (MEGENA). We propose a carefully curated set of module quality evaluation metrics to choose appropriate statistical distance-based or information theoretic similarity measures over simple linear correlation to represent the similarities between genes. Furthermore, we design and implement a stability test to ensure the robustness of the detected hub genes in the presence of noise. Finally, we propose differential module eigengene analysis for a deeper understanding of upregulation and downregulation of each module with respect to the disease and control groups for a comprehensive understanding of the clustering analysis. Besides detecting robust hub genes and modules that are supported by prior findings, we also identify previously undiscovered hub genes that can potentially lead to further biomedical research into understanding the AMD disease mechanism and developing new treatments.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript generalizes the MEGENA framework for gene co-expression network analysis on RNA-Seq data from AMD patients and controls. It introduces a curated set of module quality metrics to select statistical distance-based or information-theoretic similarity measures over linear correlation, implements a stability test to assess robustness of detected hub genes under noise, and performs differential module eigengene analysis to evaluate upregulation or downregulation relative to disease status. The central claim is that this yields both previously reported hub genes/modules supported by prior literature and novel hub genes with potential for further AMD research.
Significance. If the robustness claims hold after validation, the work could contribute a practical extension of MEGENA for noisy RNA-Seq data and help prioritize new candidate genes for AMD mechanism studies and therapies. The stability test and differential eigengene components are constructive additions that address common limitations in co-expression analyses.
major comments (3)
- [Methods (module quality evaluation)] Methods, module quality metrics subsection: The curation process for selecting similarity measures is presented without calibration against simulated networks containing known ground-truth modules; this is load-bearing for the claim that the metrics reliably identify biologically meaningful structures rather than technical artifacts in RNA-Seq co-expression.
- [Results (hub gene identification)] Results, hub gene and module reporting: No direct comparison to baseline methods such as standard WGCNA is performed on the identical AMD dataset, leaving the asserted robustness of the generalized MEGENA pipeline unbenchmarked against established alternatives.
- [Methods (stability test)] Methods, stability test description: The stability test lacks reported controls (e.g., label permutation, batch-effect injection, or noise-injection experiments) to demonstrate that it systematically rejects spurious modules arising from RNA-Seq technical variation; without these, the 'robust' designation for both known and novel hubs rests on unverified assumptions.
minor comments (2)
- [Abstract] Abstract: The phrase 'carefully curated set of module quality evaluation metrics' is used without a one-sentence summary of the metrics themselves; adding this would improve readability for readers who do not immediately consult the Methods.
- [Figures] Figure legends (throughout): Several panels lack explicit indication of which similarity measure or stability cutoff was applied; consistent annotation would aid reproducibility.
Simulated Author's Rebuttal
We thank the referee for their constructive comments, which highlight important areas for strengthening the validation of our generalized MEGENA approach. We address each major comment below and will make the indicated revisions.
read point-by-point responses
-
Referee: Methods, module quality metrics subsection: The curation process for selecting similarity measures is presented without calibration against simulated networks containing known ground-truth modules; this is load-bearing for the claim that the metrics reliably identify biologically meaningful structures rather than technical artifacts in RNA-Seq co-expression.
Authors: We agree that calibration against simulated networks with ground-truth modules would provide stronger empirical support for the module quality metrics. In the revised manuscript, we will add a dedicated simulation study in the Methods section. This will use synthetic networks with planted modules and noise levels mimicking RNA-Seq technical variation to demonstrate that the curated metrics recover true structures more reliably than alternatives, directly addressing the concern about distinguishing biological signal from artifacts. revision: yes
-
Referee: Results, hub gene and module reporting: No direct comparison to baseline methods such as standard WGCNA is performed on the identical AMD dataset, leaving the asserted robustness of the generalized MEGENA pipeline unbenchmarked against established alternatives.
Authors: We acknowledge that a head-to-head comparison on the same dataset would better substantiate the advantages of the generalized MEGENA pipeline. We will add this analysis to the Results section, applying standard WGCNA to the identical AMD RNA-Seq data and comparing outputs on metrics including module quality scores, overlap with previously reported AMD genes, hub gene stability, and differential eigengene patterns. revision: yes
-
Referee: Methods, stability test description: The stability test lacks reported controls (e.g., label permutation, batch-effect injection, or noise-injection experiments) to demonstrate that it systematically rejects spurious modules arising from RNA-Seq technical variation; without these, the 'robust' designation for both known and novel hubs rests on unverified assumptions.
Authors: We agree that explicit controls are necessary to validate that the stability test rejects technical artifacts. We will expand the Methods to include label-permutation tests establishing significance thresholds above chance levels and controlled noise-injection experiments simulating RNA-Seq batch effects and technical variation. Results from these controls will be reported to confirm the robustness of both known and novel hub genes. revision: yes
Circularity Check
No circularity: analysis applies external MEGENA framework to independent RNA-Seq data with added but non-self-referential metrics
full rationale
The paper generalizes an existing method (MEGENA) by proposing module quality metrics to select similarity measures, adding a stability test, and performing differential eigengene analysis on real AMD/control RNA-Seq data. No equations, definitions, or steps reduce outputs (hub genes, modules) to inputs by construction. Module selection uses data-driven metrics on external observations rather than fitting parameters that are then renamed as predictions. No load-bearing self-citations or uniqueness theorems imported from prior author work appear in the derivation chain. The central claims rest on application to independent data, not tautological re-expression of fitted values.
Axiom & Free-Parameter Ledger
free parameters (2)
- Similarity measure selection via curated quality metrics
- Module detection thresholds and stability cutoffs
axioms (1)
- domain assumption Genes with coordinated expression patterns participate in related biological processes
Reference graph
Works this paper leans on
-
[1]
Retinal transcrip- tome and eQTL analyses identify genes associated with age-related macular degeneration,
R. Ratnapriya, O. A. Sosina, M. R. Starostiket al., “Retinal transcrip- tome and eQTL analyses identify genes associated with age-related macular degeneration,”Nature Genetics, vol. 51, no. 4, pp. 606–610, Apr. 2019
2019
-
[2]
Age-related macular degenera- tion—clinical review and genetics update,
R. Ratnapriya and E. Y . Chew, “Age-related macular degenera- tion—clinical review and genetics update,”Clinical Genetics, vol. 84, no. 2, pp. 160–166, 2013
2013
-
[3]
Genome-wide association analyses identify distinct genetic architectures for age-related macular degeneration across ancestries,
B. R. Gorman, G. V oloudakis, R. P. Igoet al., “Genome-wide association analyses identify distinct genetic architectures for age-related macular degeneration across ancestries,”Nature Genetics, vol. 56, no. 12, pp. 2659–2671, Dec. 2024
2024
-
[4]
Age-Related Macular Degeneration: Genetics and Biology Coming Together,
L. G. Fritsche, R. N. Farisset al., “Age-Related Macular Degeneration: Genetics and Biology Coming Together,”Annual Review of Genomics and Human Genetics, vol. 15, no. 1, pp. 151–171, Aug. 2014
2014
-
[5]
Inte- grating explainable machine learning and transcriptomics data reveals cell-type specific immune signatures underlying macular degeneration,
K. Ma, H. Nakajima, N. Basak, A. Barman, and R. Ratnapriya, “Inte- grating explainable machine learning and transcriptomics data reveals cell-type specific immune signatures underlying macular degeneration,” npj Genomic Medicine, vol. 10, no. 1, p. 48, Jun. 2025, publisher: Nature Publishing Group
2025
-
[6]
The Role of Gene Expression Regulation on Genetic Risk of Age-Related Macular Degeneration,
R. Ratnapriya, “The Role of Gene Expression Regulation on Genetic Risk of Age-Related Macular Degeneration,” inRetinal Degenerative Diseases XIX, J. D. Ash, E. Pierce, R. E. Anderson, C. Bowes Rickman, J. G. Hollyfield, and C. Grimm, Eds. Cham: Springer International Publishing, 2023, vol. 1415, pp. 61–66, series Title: Advances in Experimental Medicine a...
2023
-
[7]
A graphical method for identifying gene clusters from rna sequencing data,
J. R. Patock, R. Ratnapriya, and A. Barman, “A graphical method for identifying gene clusters from rna sequencing data,” in2025 IEEE International Conference on Data Mining Workshops (ICDMW), 2025, pp. 485–492
2025
-
[8]
Bioinformatics perspectives on transcriptomics: A comprehensive re- view of bulk and single-cell RNA sequencing analyses,
J. A. Tzec-Interi ´an, D. Gonz ´alez-Padilla, and E. B. G ´ongora-Castillo, “Bioinformatics perspectives on transcriptomics: A comprehensive re- view of bulk and single-cell RNA sequencing analyses,”Quantitative Biology, vol. 13, no. 2, p. e78, 2025
2025
-
[9]
Transcriptome profiling and co-expression network analysis of lncRNAs and mRNAs in colorectal cancer by RNA sequencing,
M. Li, D. Guo, X. Chen, X. Lu, X. Huang, and Y . Wu, “Transcriptome profiling and co-expression network analysis of lncRNAs and mRNAs in colorectal cancer by RNA sequencing,”BMC Cancer, vol. 22, no. 1, p. 780, Jul. 2022
2022
-
[10]
hdWGCNA identifies co-expression networks in high-dimensional transcriptomics data,
S. Morabito, F. Reese, N. Rahimzadehet al., “hdWGCNA identifies co-expression networks in high-dimensional transcriptomics data,”Cell Reports Methods, vol. 3, no. 6, p. 100498, Jun. 2023
2023
-
[11]
Gene Co-expression Network Analysis,
J. D. Montenegro, “Gene Co-expression Network Analysis,”Methods in Molecular Biology, vol. 2443, pp. 387–404, 2022, place: Clifton, N.J
2022
-
[12]
Identification of the Hub Genes in Alzheimer’s Disease,
H. Gui, Q. Gong, J. Jiang, M. Liu, and H. Li, “Identification of the Hub Genes in Alzheimer’s Disease,”Computational and Mathematical Methods in Medicine, vol. 2021, no. 1, p. 6329041, 2021
2021
-
[13]
Gene expression patterns combined with network analysis identify hub genes associated with bladder cancer,
D. Bi, H. Ning, S. Liuet al., “Gene expression patterns combined with network analysis identify hub genes associated with bladder cancer,” Computational Biology and Chemistry, vol. 56, pp. 71–83, Jun. 2015
2015
-
[14]
Predicting Hub Genes Asso- ciated with Cervical Cancer through Gene Co-Expression Networks,
S.-P. Deng, L. Zhu, and D.-S. Huang, “Predicting Hub Genes Asso- ciated with Cervical Cancer through Gene Co-Expression Networks,” IEEE/ACM Transactions on Computational Biology and Bioinformatics, vol. 13, no. 1, pp. 27–35, Jan. 2016
2016
-
[15]
WGCNA: an R package for weighted correlation network analysis,
P. Langfelder and S. Horvath, “WGCNA: an R package for weighted correlation network analysis,”BMC Bioinformatics, vol. 9, no. 1, p. 559, Dec. 2008
2008
-
[16]
Multiscale Embedded Gene Co-expression Network Analysis,
W.-M. Song and B. Zhang, “Multiscale Embedded Gene Co-expression Network Analysis,”PLOS Computational Biology, vol. 11, no. 11, p. e1004574, Nov. 2015, publisher: Public Library of Science
2015
-
[17]
Building complex networks with Platonic solids,
W.-M. Song, T. Di Matteo, and T. Aste, “Building complex networks with Platonic solids,”Physical review. E, Statistical, nonlinear, and soft matter physics, vol. 85, p. 046115, Apr. 2012
2012
-
[18]
Cell-type-specific co-expression inference from single cell RNA-sequencing data,
C. Su, Z. Xu, X. Shan, B. Cai, H. Zhao, and J. Zhang, “Cell-type-specific co-expression inference from single cell RNA-sequencing data,”Nature Communications, vol. 14, no. 1, p. 4846, Aug. 2023, publisher: Nature Publishing Group
2023
-
[19]
Eigengene networks for studying the relationships between co-expression modules,
P. Langfelder and S. Horvath, “Eigengene networks for studying the relationships between co-expression modules,”BMC Systems Biology, vol. 1, no. 1, p. 54, Nov. 2007
2007
-
[20]
Quiescent stem cell marker genes in glioma gene networks are sufficient to distinguish between normal and glioblastoma (GBM) samples,
S. Mukherjee, “Quiescent stem cell marker genes in glioma gene networks are sufficient to distinguish between normal and glioblastoma (GBM) samples,”Scientific Reports, vol. 10, no. 1, p. 10937, Jul. 2020
2020
-
[21]
Identification of molecular signatures associated with sleep disorder and Alzheimer’s disease,
L. Liang, J. Yan, X. Huang, C. Zou, L. Chen, R. Li, J. Xie, M. Pan, D. Zou, and Y . Liu, “Identification of molecular signatures associated with sleep disorder and Alzheimer’s disease,”Frontiers in Psychiatry, vol. 13, p. 925012, 2022
2022
-
[22]
Brain and blood transcriptome profiles delineate common genetic pathways across suicidal ideation and suicide,
S. Sun, Q. Liu, Z. Wang, Y .-Y . Huang, M. E. Sublette, A. J. Dwork, G. Rosoklija, Y . Ge, H. Galfalvy, J. J. Mann, and F. Haghighi, “Brain and blood transcriptome profiles delineate common genetic pathways across suicidal ideation and suicide,”Molecular Psychiatry, vol. 29, no. 5, pp. 1417–1426, May 2024
2024
-
[23]
Genetic sus- ceptibility to age-related macular degeneration: a paradigm for dissecting complex disease traits,
A. Swaroop, K. E. Branham, W. Chen, and G. Abecasis, “Genetic sus- ceptibility to age-related macular degeneration: a paradigm for dissecting complex disease traits,”Human Molecular Genetics, vol. 16, no. R2, pp. R174–R182, Jul. 2007
2007
-
[24]
Genome-wide meta-analysis identifies novel loci associated with age-related macular degeneration,
X. Han, P. Gharahkhani, P. Mitchell, G. Liew, A. W. Hewitt, and S. Mac- Gregor, “Genome-wide meta-analysis identifies novel loci associated with age-related macular degeneration,”Journal of Human Genetics, vol. 65, no. 8, pp. 657–665, Aug. 2020
2020
-
[25]
A transcriptome-wide association study based on 27 tissues identifies 106 genes potentially relevant for disease pathology in age-related macular degeneration,
T. Strunz, S. Lauwen, C. Kielet al., “A transcriptome-wide association study based on 27 tissues identifies 106 genes potentially relevant for disease pathology in age-related macular degeneration,”Scientific Reports, vol. 10, no. 1, p. 1584, Jan. 2020
2020
-
[26]
Genetic Insights into Age- Related Macular Degeneration,
n. Bhumika, N. S. Bora, and P. S. Bora, “Genetic Insights into Age- Related Macular Degeneration,”Biomedicines, vol. 12, no. 7, p. 1479, Jul. 2024
2024
-
[27]
Information theoretic measures for clusterings comparison: Variants, properties, normalization and correc- tion for chance,
N. X. Vinh, J. Epps, and J. Bailey, “Information theoretic measures for clusterings comparison: Variants, properties, normalization and correc- tion for chance,”Journal of Machine Learning Research, vol. 11, pp. 2837–2854, 2010
2010
-
[28]
A new metric for probability distributions,
D. M. Endres and J. E. Schindelin, “A new metric for probability distributions,”IEEE Transactions on Information Theory, vol. 49, no. 7, pp. 1858–1860, 2003
2003
-
[29]
Divergence measures based on the shannon entropy,
J. Lin, “Divergence measures based on the shannon entropy,”IEEE Transactions on Information Theory, vol. 37, no. 1, pp. 145–151, 1991
1991
-
[30]
A tool for filtering information in complex systems,
M. Tumminello, T. Aste, T. Di Matteo, and R. N. Mantegna, “A tool for filtering information in complex systems,”Proceedings of the National Academy of Sciences, vol. 102, no. 30, pp. 10 421–10 426, Jul. 2005, publisher: Proceedings of the National Academy of Sciences
2005
-
[31]
Statistical mechanics of complex net- works,
R. Albert and A.-L. Barab ´asi, “Statistical mechanics of complex net- works,”Reviews of Modern Physics, vol. 74, no. 1, pp. 47–97, Jan. 2002, publisher: American Physical Society
2002
-
[32]
Similarity index based on local paths for link prediction of complex networks,
L. L ¨u, C.-H. Jin, and T. Zhou, “Similarity index based on local paths for link prediction of complex networks,”Physical Review E, vol. 80, no. 4, p. 046122, Oct. 2009, publisher: American Physical Society
2009
-
[33]
Modularity and community structure in networks,
M. E. J. Newman, “Modularity and community structure in networks,” Proceedings of the National Academy of Sciences of the United States of America, vol. 103, no. 23, pp. 8577–8582, Jun. 2006
2006
-
[34]
Defining and evaluating network communities based on ground-truth,
J. Yang and J. Leskovec, “Defining and evaluating network communities based on ground-truth,” inProceedings of the ACM SIGKDD Workshop on Mining Data Semantics, ser. MDS ’12. New York, NY , USA: Association for Computing Machinery, Aug. 2012, pp. 1–8
2012
-
[35]
Integrated transcriptomic analysis of human induced pluripotent stem cell-derived osteogenic differentiation reveals a regulatory role of KLF16,
Y . Ru, M. Ma, X. Zhouet al., “Integrated transcriptomic analysis of human induced pluripotent stem cell-derived osteogenic differentiation reveals a regulatory role of KLF16,”bioRxiv: The Preprint Server for Biology, p. 2024.02.11.579844, Jan. 2025
2024
-
[36]
Complement in age- related macular degeneration: a focus on function,
D. T. Bradley, P. F. Zipfel, and A. E. Hughes, “Complement in age- related macular degeneration: a focus on function,”Eye, vol. 25, no. 6, pp. 683–693, Jun. 2011
2011
-
[37]
Association between the SERPING1 gene and age-related macular degeneration: a two-stage case–control study,
S. Ennis, C. Jomaryet al., “Association between the SERPING1 gene and age-related macular degeneration: a two-stage case–control study,” The Lancet, vol. 372, no. 9652, pp. 1828–1834, Nov. 2008
2008
-
[38]
Pleiotropic effects of all-trans retinoic acid in attenuating the hallmarks of colorectal cancer- challenges and scope of differentiation therapy,
A. A. Michael, P. Balakrishnan, P. Tamizhmani, K. Thirunavukarasu, S. Subramaniam, and T. Velusamy, “Pleiotropic effects of all-trans retinoic acid in attenuating the hallmarks of colorectal cancer- challenges and scope of differentiation therapy,”Cancer Gene Therapy, Dec. 2025
2025
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.