Recognition: unknown
Towards Accelerated SCF Workflows with Equivariant Density-Matrix Learning and Analytic Refinement
Pith reviewed 2026-05-07 08:22 UTC · model grok-4.3
The pith
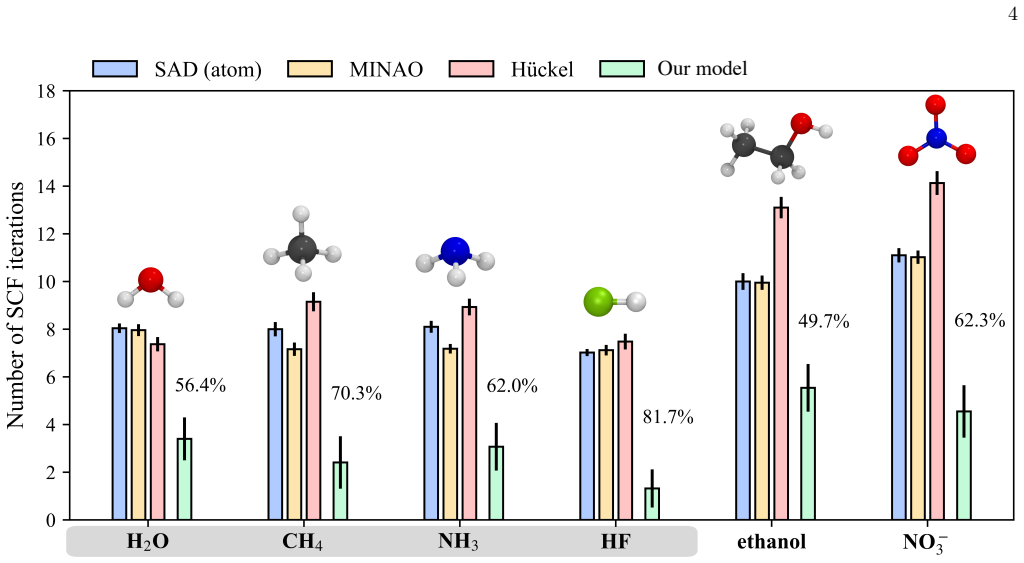
An equivariant neural network predicts one-electron density matrices that accelerate SCF calculations by 49 to 81 percent on small molecules.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
dm-PhiSNet is a PhiSNet-based equivariant model that predicts 1-RDMs directly from molecular geometries in an AO basis. A two-stage training schedule introduces progressively stronger physical objectives, and the output is passed through an analytic refinement block that restores electron-number conservation, drives the matrix toward generalized idempotency in the AO metric, and regularizes the occupation spectrum of the Löwdin-orthogonalized density. On H2O, CH4, NH3, HF, ethanol, and NO3-, the resulting 1-RDMs reduce SCF iteration counts by 49-81 percent relative to standard initializations while also producing accurate one-shot total energies and Hellmann-Feynman atomic forces without any
What carries the argument
dm-PhiSNet, an equivariant PhiSNet architecture that outputs atomic-orbital-basis one-electron reduced density matrices, paired with a two-stage training schedule and an analytic refinement block that enforces electron conservation, generalized idempotency, and Löwdin occupation regularization.
If this is right
- SCF calculations can begin from the learned 1-RDM instead of superposition-of-atomic-densities or core-Hamiltonian guesses, shortening wall-clock time for single-point and geometry-optimization jobs.
- One-shot total energies and Hellmann-Feynman forces extracted from the refined 1-RDM become usable for rapid property screening without completing the full iterative SCF cycle.
- The same pipeline supplies density matrices that already satisfy basic physical constraints, reducing the risk of convergence failures that plague purely data-driven guesses.
- Existing quantum-chemistry codes can incorporate the model as a drop-in pre-step to accelerate workflows that repeatedly solve the SCF problem.
Where Pith is reading between the lines
- If the refinement block proves robust, the method could be inserted into molecular-dynamics loops to supply updated electronic structure at lower cost than full SCF at every step.
- Extending the same architecture to larger or open-shell molecules would test whether equivariant density-matrix learning scales beyond the closed-shell regime studied here.
- The observation that force accuracy emerges without force supervision suggests that density-matrix supervision alone may be sufficient to capture chemically meaningful electronic structure for many properties.
Load-bearing premise
The two-stage training schedule and analytic refinement block, shown only on six small closed-shell molecules, will produce reliable, solver-ready 1-RDM initial guesses for chemically diverse systems without introducing new instabilities or requiring system-specific retuning.
What would settle it
Running the trained model on an open-shell or transition-metal system and finding that the predicted 1-RDM either fails to reduce SCF steps below standard guesses or produces divergent or unstable SCF behavior would falsify the claim that the method supplies general solver-ready initializations.
Figures


read the original abstract
We present \textsc{dm-PhiSNet}, a physically constrained \textsc{PhiSNet}-based equivariant model that predicts one-electron reduced density matrices (1-RDMs) directly from molecular geometries in an atomic-orbital (AO) basis for accelerated self-consistent field (SCF) workflows. Training follows a two-stage schedule with progressively introduced physically motivated objectives, and the resulting predictions are refined by a lightweight analytic block. This block enforces electron-number conservation, drives the 1-RDM toward generalized idempotency in the AO metric, and regularizes the occupation spectrum of the L\"owdin-orthogonalized density. Across six closed-shell systems -- H$_2$O, CH$_4$, NH$_3$, HF, ethanol, and NO$_3^-$ -- the refined 1-RDMs provide SCF initial guesses that substantially reduce iteration steps by 49--81\% relative to standard initializations. Beyond SCF acceleration, the learned 1-RDMs yield accurate one-shot total energies and Hellmann--Feynman atomic forces without force supervision, indicating that the model captures chemically meaningful electronic structure. These results demonstrate that combining equivariant learning with analytic constraint enforcement provides a simple, general route to solver-ready density-matrix initializations and accelerated SCF workflows.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript introduces dm-PhiSNet, an equivariant PhiSNet-based model that directly predicts one-electron reduced density matrices (1-RDMs) in an atomic-orbital basis from molecular geometries. Predictions undergo a two-stage training schedule followed by a lightweight analytic refinement block that enforces electron-number conservation, generalized idempotency in the AO metric, and Löwdin-orthogonalized occupation regularization. On the six closed-shell systems H2O, CH4, NH3, HF, ethanol, and NO3-, the refined 1-RDMs are shown to serve as SCF initial guesses that reduce iteration counts by 49-81% relative to standard initializations while also delivering accurate one-shot total energies and Hellmann-Feynman forces without any force supervision.
Significance. If the approach proves robust beyond the current test set, the combination of equivariant density-matrix learning with independent analytic constraints could meaningfully accelerate routine SCF workflows and enable direct extraction of chemically meaningful forces from geometry-only predictions. The absence of force supervision and the use of physically derived refinement steps are notable strengths that distinguish this from purely data-driven initial-guess methods.
major comments (2)
- [Abstract] Abstract: The claim of a 'simple, general route' to solver-ready 1-RDM initial guesses rests on results from only six small closed-shell molecules. No data are provided for open-shell, transition-metal, or larger systems where the raw network prediction may lie farther from the physical manifold; this directly affects whether the analytic refinement block remains stable and whether the reported 49-81% iteration savings and force accuracy generalize.
- [Abstract] Abstract and methods: No information is given on training-set size, chemical diversity, train/validation/test splits, or statistical error bars on the iteration counts and energy/force errors. These omissions make it impossible to judge the robustness of the numerical gains that underpin the central SCF-acceleration claim.
minor comments (1)
- [Abstract] Notation for the 1-RDM and its refined version should be introduced once and used consistently; the current abstract alternates between 'predicted 1-RDMs' and 'refined 1-RDMs' without a clear distinction in the first paragraph.
Simulated Author's Rebuttal
We thank the referee for their constructive and detailed report. We address each major comment point by point below, indicating where revisions have been made to the manuscript.
read point-by-point responses
-
Referee: [Abstract] Abstract: The claim of a 'simple, general route' to solver-ready 1-RDM initial guesses rests on results from only six small closed-shell molecules. No data are provided for open-shell, transition-metal, or larger systems where the raw network prediction may lie farther from the physical manifold; this directly affects whether the analytic refinement block remains stable and whether the reported 49-81% iteration savings and force accuracy generalize.
Authors: We agree that the evaluation is restricted to six small closed-shell molecules and that this limits the strength of any claim to a fully general route. The analytic refinement block relies on system-independent physical constraints (electron-number conservation, generalized idempotency, and Löwdin occupation regularization), which in principle should remain stable, but we have no evidence for its behavior when the raw network prediction deviates more substantially from the physical manifold. In the revised manuscript we have removed the phrase 'simple, general route' from the abstract, replaced it with language that explicitly qualifies the scope to the systems studied, and added a new limitations paragraph in the Discussion section that outlines the need for future tests on open-shell, transition-metal, and larger molecules. revision: yes
-
Referee: [Abstract] Abstract and methods: No information is given on training-set size, chemical diversity, train/validation/test splits, or statistical error bars on the iteration counts and energy/force errors. These omissions make it impossible to judge the robustness of the numerical gains that underpin the central SCF-acceleration claim.
Authors: We apologize for the lack of these details in the abstract and the high-level methods description. The full manuscript already contains the relevant information in the Methods section (dataset generation via ab initio molecular dynamics, train/validation/test splits, and chemical composition of the six closed-shell species). To address the referee's concern directly, we have added a concise summary of training-set sizes, splits, and chemical diversity to both the abstract and the opening of the Methods section in the revision. We have also recomputed and reported statistical error bars (standard deviations across independent training runs) on all iteration counts, energy errors, and force errors in the revised figures and tables. revision: yes
- Performance and stability of the analytic refinement block on open-shell, transition-metal, or substantially larger molecular systems, for which no data are available in the current work.
Circularity Check
No significant circularity in the 1-RDM prediction and analytic refinement chain
full rationale
The paper's workflow trains an equivariant neural network (dm-PhiSNet) to predict 1-RDMs directly from molecular geometries using a two-stage schedule incorporating physically motivated objectives, then applies an independent analytic refinement block enforcing electron-number conservation, AO-metric idempotency, and Löwdin occupation regularization. These constraints derive from standard quantum mechanics rather than from the network outputs or fitted parameters themselves. No step reduces by construction to its inputs: the neural predictions are data-driven approximations, the refinement is a post-processing projection onto the physical manifold, and the reported SCF iteration reductions (49-81%) and one-shot energy/force accuracies are empirical measurements on the six closed-shell systems rather than tautological redefinitions. No load-bearing self-citations, uniqueness theorems, or ansatzes smuggled via prior work are invoked in the provided derivation; the central claim remains an independent combination of learning and analytic enforcement.
Axiom & Free-Parameter Ledger
free parameters (2)
- loss weighting schedule
- refinement regularization strength
axioms (2)
- domain assumption Molecular Hamiltonians and observables are equivariant under rotations and translations
- domain assumption A valid 1-RDM must conserve electron number and be approximately idempotent in the AO metric
Reference graph
Works this paper leans on
-
[1]
Westermayr, K
J. Westermayr, K. T. Sch¨ utt, O. A. von Lilienfeld, and R. J. Maurer, J. Chem. Phys.154, 230903 (2021)
2021
-
[2]
Chandrasekaran, D
A. Chandrasekaran, D. Kamal, R. Batra, C. Kim, L.-Q. Chen, and R. Ramprasad, npj Comput. Mater.5, 22 (2019)
2019
-
[3]
Hu and W
X. Hu and W. Yang, J. Chem. Phys.132, 054109 (2010)
2010
-
[4]
Canc` es and C
E. Canc` es and C. Le Bris,On the convergence of SCF algorithms for the Hartree–Fock equations, Tech. Rep. CERMICS-99-184 (CERMICS, `Ecole Nationale des Ponts et Chauss´ ees, 1999) preprint/technical report (widely cited in later SCF convergence analyses)
1999
-
[5]
D. A. Mazziotti,Reduced-Density-Matrix Mechanics: With Application to Many-Electron Atoms and Molecules (Wiley, 2007)
2007
-
[6]
L¨ owdin, J
P.-O. L¨ owdin, J. Chem. Phys.18, 365 (1950). 7
1950
-
[7]
K. T. Sch¨ utt, M. Gastegger, A. Tkatchenko, K.-R. M¨ uller, and R. J. Maurer, Nat. Commun.10, 5024 (2019)
2019
-
[8]
Grisafi, A
A. Grisafi, A. Fabrizio, B. Meyer, D. M. Wilkins, M. Ce- riotti, and C. Corminboeuf, ACS Cent. Sci.5, 57 (2019)
2019
-
[9]
A. M. Lewis, A. Grisafi, M. Ceriotti, and M. Rossi, J. Chem. Theory Comput.17, 7203 (2021)
2021
-
[10]
Grisafi and M
A. Grisafi and M. Ceriotti, J. Chem. Theory Comput. 19, 337 (2023)
2023
-
[11]
K. Ryczko, K. Mills, I. Luchak, C. Homenick, and I. Tamblyn, “Deep neural network computes elec- tron densities and energies of a large set of organic molecules faster than density functional theory,” (2018), arXiv:1809.02723 [physics.chem-ph]
-
[12]
Del Rio and R
B. Del Rio and R. Ramprasad, npj Comput. Mater.9, 68 (2023)
2023
-
[13]
J. C. Snyder, M. Rupp, K. Hansen, K.-R. M¨ uller, and K. Burke, Phys. Rev. Lett.108, 253002 (2012)
2012
-
[14]
Bogojeski, L
M. Bogojeski, L. Vogt-Maranto, M. E. Tuckerman, K.-R. M¨ uller, and K. Burke, Nat. Commun.11, 5223 (2020)
2020
-
[15]
Pederson, B
R. Pederson, B. Kalita, and K. Burke, Nature Reviews Physics4, 357 (2022)
2022
-
[16]
Focassio, F
B. Focassio, F. A. Faber, and O. A. von Lilienfeld, npj Comput. Mater.9, 42 (2023)
2023
-
[17]
H. Li, Z. Wang, N. Zou, M. Ye, R. Xu, X. Gong, W. Duan, and Y. Xu, Nat. Comput. Sci.2, 367 (2022)
2022
-
[18]
X. Gong, H. Li, N. Zou, R. Xu, W. Duan, and Y. Xu, Nat. Commun.14, 2848 (2023)
2023
-
[19]
Zhang, J
L. Zhang, J. Han, H. Wang, R. Car, and W. E, npj Comput. Mater.8, 26 (2022)
2022
-
[20]
Zhong, Y
Y. Zhong, Y. Li, Z. Xu, X. Chen, and L. Zhang, npj Comput. Mater.9, 48 (2023)
2023
-
[21]
S. Gong, Y. Li, H. Wang, and L. Zhang, Nat. Commun. 14, 3589 (2023)
2023
-
[22]
Many- body message passing for equivariant prediction of elec- tronic hamiltonians,
T. Qian, Y. Ma, J. Chen, and L. Zhang, “Many- body message passing for equivariant prediction of elec- tronic hamiltonians,” (2025), arXiv:2508.15108 [cond- mat.mtrl-sci]
-
[23]
H. Liu, S. Guan, Z. Wang, Z. Zhou, Z. Qu, and W. Liu, JACS Au (2025), 10.1021/jacsau.5c01200, early View
-
[24]
A fully differ- entiable framework for machine learning quantum chem- istry,
A. Suman, R. Jain, and R. Ramprasad, “A fully differ- entiable framework for machine learning quantum chem- istry,” (2025), arXiv:2504.01187 [physics.chem-ph]
-
[25]
O. T. Unke, M. Bogojeski, M. Gastegger, M. Geiger, T. Smidt, and K.-R. M¨ uller, inAdv. Neural Inf. Pro- cess. Syst., Vol. 34 (2021) pp. 14434–14447
2021
-
[26]
H. Shao, L. Zhang, and H. Wang, Nat. Commun.14, 6294 (2023)
2023
-
[27]
Hazra, U
S. Hazra, U. Patil, and S. Sanvito, J. Chem. Theory Comput.20, 4569 (2024)
2024
-
[28]
B. Rana, N. Viot, J. A. Martinez B., X. Shao, P. Ramos, and M. Pavanello, J. Chem. Theory Comput.20, 4357 (2024)
2024
-
[29]
Improving density matrix electronic struc- ture method by deep learning,
Z. Tang, Z. Nianlong, H. Li, Y. Wang, Z. Yuan, H. Tao, Y. Li, Z. Chen, B. Zhao, M. Sun, H. Jiang, W. Duan, and Y. Xu, “Improving density matrix electronic struc- ture method by deep learning,” (2024), arXiv:2406.17561 [physics.chem-ph]
-
[30]
McWeeny, Rev
R. McWeeny, Rev. Mod. Phys.32, 335 (1960)
1960
-
[31]
Q. Sun, X. Zhang, S. Banerjee, P. Bao, M. Barbry, N. S. Blunt, N. A. Bogdanov, G. H. Booth, J. Chen, Z.-H. Cui, et al., J. Chem. Phys.153, 024109 (2020)
2020
-
[32]
Weigend and R
F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys. 7, 3297 (2005)
2005
-
[33]
Rappoport and F
D. Rappoport and F. Furche, J. Chem. Phys.133, 134105 (2010)
2010
-
[34]
Chmiela, A
S. Chmiela, A. Tkatchenko, H. E. Sauceda, I. Poltavsky, K. T. Sch¨ utt, and K.-R. M¨ uller, Sci. Adv.3, e1603015 (2017)
2017
-
[35]
Hjorth Larsen, J
A. Hjorth Larsen, J. J. Mortensen, J. Blomqvist, I. E. Castelli, R. Christensen, M. Du lak, J. Friis, M. N. Groves, B. Hammer, C. Hargus, E. D. Hermes, P. C. Jennings, P. B. Jensen, J. Kermode, J. R. Kitchin, E. L. Kolsbjerg, J. Kubal, K. Kaasbjerg, S. Lysgaard, J. B. Maronsson, T. Maxson, T. Olsen, L. Pastewka, A. Pe- terson, C. Rostgaard, J. Schiøtz, O....
2017
-
[36]
V. Blum, R. Gehrke, F. Hanke, P. Havu, V. Havu, X. Ren, K. Reuter, and M. Scheffler, Comput. Phys. Commun.180, 2175 (2009)
2009
-
[37]
Bannwarth, S
C. Bannwarth, S. Ehlert, and S. Grimme, J. Chem. The- ory Comput.15, 1652 (2019)
2019
-
[38]
Alml¨ of, K
J. Alml¨ of, K. Faegri, and K. Korsell, J. Comput. Chem. 3, 385 (1982)
1982
-
[39]
J. H. Van Lenthe, R. Zwaans, H. J. J. Van Dam, and M. F. Guest, J. Comput. Chem.27, 926 (2006)
2006
-
[40]
H¨ uckel, Z
E. H¨ uckel, Z. Phys.70, 204 (1931)
1931
-
[41]
H¨ uckel, Z
E. H¨ uckel, Z. Phys.72, 310 (1931)
1931
-
[42]
Pulay, Chem
P. Pulay, Chem. Phys. Lett.73, 393 (1980)
1980
-
[43]
Pulay, J
P. Pulay, J. Comput. Chem.3, 556 (1982)
1982
-
[44]
P.-F. Loos, N. Galland, and D. Jacquemin, J. Phys. Chem. Lett.9, 4646 (2018)
2018
-
[45]
Hellmann,Einf¨ uhrung in die Quantenchemie(Franz Deuticke, Leipzig, 1937)
H. Hellmann,Einf¨ uhrung in die Quantenchemie(Franz Deuticke, Leipzig, 1937)
1937
-
[46]
R. P. Feynman, Phys. Rev.56, 340 (1939)
1939
-
[47]
O. T. Unke, S. Chmiela, M. Gastegger, K. T. Sch¨ utt, H. E. Sauceda, and K.-R. M¨ uller, Nat. Commun.12, 7273 (2021)
2021
-
[48]
Batzner, A
S. Batzner, A. Musaelian, L. Sun, M. Geiger, J. P. Mailoa, M. Kornbluth, N. Molinari, T. E. Smidt, and B. Kozinsky, Nat. Commun.13, 2453 (2022)
2022
-
[49]
I. Batatia, D. P. Kov´ acs, G. N. C. Simm, C. Ortner, and G. Cs´ anyi, “Mace: Higher order equivariant mes- sage passing neural networks for fast and accurate force fields,” (2022), arXiv:2206.07697
-
[50]
L. C´ azares-Trejo, M. Loreto-Silva, and H. E. Sauceda, “Delta-learned force fields for nonbonded interactions: Addressing the strength mismatch between covalent- nonbonded interaction for global models,” (2025), arXiv:2511.01913 [physics.chem-ph]
-
[51]
O. T. Unke, S. Chmiela, H. E. Sauceda, M. Gastegger, I. Poltavsky, K. T. Sch¨ utt, A. Tkatchenko, and K.-R. M¨ uller, Chem. Rev.121, 10142 (2021)
2021
-
[52]
Car and M
R. Car and M. Parrinello, Phys. Rev. Lett.55, 2471 (1985)
1985
-
[53]
National Research Council,Mathematical Challenges from Theoretical/Computational Chemistry(The Na- tional Academies Press, Washington, DC, 1995)
1995
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.