Recognition: unknown
VibroML: an automated toolkit for high-throughput vibrational analysis and dynamic instability remediation of crystalline materials using machine-learned potentials
Pith reviewed 2026-05-07 06:25 UTC · model grok-4.3
The pith
VibroML uses an energy-guided genetic algorithm on machine-learned potentials to automatically remediate dynamical instabilities and uncover stable crystal polymorphs.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
VibroML couples machine-learned interatomic potentials with an energy-guided genetic algorithm that navigates the potential energy surface to identify diverse dynamically stable polymorphs for high-symmetry structures showing instabilities, includes molecular dynamics to confirm finite-temperature retention, and integrates with combinatorial prediction to stabilize frustrated topologies through targeted alloying, as shown by rescuing perovskite networks and mining databases for abandoned quaternary and quinary stoichiometries.
What carries the argument
Energy-guided genetic algorithm driven by machine-learned interatomic potentials that explores the potential energy surface to locate diverse dynamically stable polymorphs.
If this is right
- Enables stabilization of functional perovskite networks such as Cs2KInI6 and KTaSe3 through targeted alloying.
- Recovers dynamically stable low-symmetry candidates from databases where roughly half of quaternary and nearly all quinary elemental combinations lack any structural entries.
- Moves high-throughput workflows from instability detection alone to full structural remediation plus thermal validation.
- Supports systematic exploration of complex compositions that standard pipelines leave abandoned.
Where Pith is reading between the lines
- The method could lower the fraction of computationally generated materials that are discarded solely because of initial instabilities, opening routes to more candidates for applications.
- Pairing the remediation step with additional prediction engines might help close coverage gaps in materials databases for multi-element systems.
- The emphasis on finite-temperature checks suggests that purely harmonic zero-kelvin analyses may systematically miss viable structures in future screening campaigns.
Load-bearing premise
The machine-learned potentials must represent the true potential energy surface accurately enough for the genetic algorithm to reach genuine stable polymorphs instead of metastable or spurious minima.
What would settle it
Recomputing the phonon spectra and running extended molecular dynamics on the remediated polymorphs with density functional theory, then checking against any available experimental stability data, would confirm or refute whether the structures are truly stable.
Figures



read the original abstract
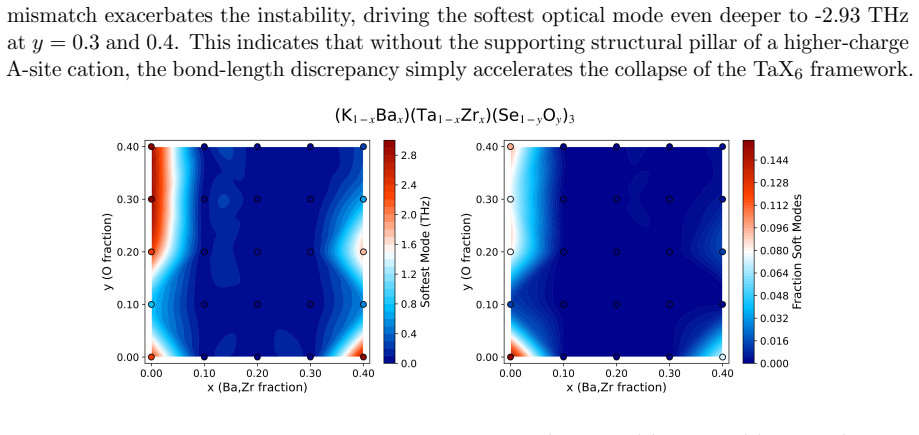
While machine-learned interatomic potentials (MLIPs) accelerate phonon dispersion calculations, merely identifying dynamical instabilities in computationally predicted materials is insufficient; automated pathways to resolve them are required. We introduce VibroML, an open-source Python toolkit driven by foundational MLIPs that shifts the paradigm from stability verification to automated structural remediation. VibroML employs an energy-guided genetic algorithm that vastly outperforms traditional soft-mode following, efficiently navigating the potential energy surface to uncover diverse, dynamically stable polymorphs. As 0 K harmonic stability does not guarantee macroscopic viability, an automated molecular dynamics workflow evaluates finite-temperature structural retention. VibroML also couples with ProtoCSP, our combinatorial structure prediction engine, to stabilize frustrated crystal topologies via targeted alloying, successfully rescuing functional perovskite networks like Cs$_2$KInI$_6$ and KTaSe$_3$. Demonstrating broader applicability, we mined the Alexandria database -- where ~50% of quaternary and 99.5% of quinary elemental combinations lack any structural entries -- to identify thousands of abandoned, high-symmetry stoichiometries. Deploying ProtoCSP's "cold start" retrieval and VibroML's evolutionary search on a sample, we successfully identified dynamically stable low-symmetry candidates. Through integrated structural remediation, thermal validation, and systematic compositional exploration, VibroML enables a comprehensive deep-screening approach, yielding physically sound structural propositions that far surpass standard high-throughput workflows.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript introduces VibroML, an open-source Python toolkit for high-throughput vibrational analysis and dynamic instability remediation in crystalline materials using foundational machine-learned interatomic potentials (MLIPs). It employs an energy-guided genetic algorithm claimed to outperform traditional soft-mode following for discovering diverse, dynamically stable polymorphs, incorporates automated molecular dynamics workflows for finite-temperature structural retention, and integrates with ProtoCSP for targeted alloying to stabilize frustrated topologies. Applications include remediation of specific perovskites (Cs₂KInI₆ and KTaSe₃) and mining the Alexandria database to identify stable low-symmetry candidates from high-symmetry stoichiometries lacking prior entries.
Significance. If the central claims hold with proper validation, VibroML could represent a practical advance in automated materials screening by shifting focus from instability detection to remediation, potentially improving efficiency in high-throughput workflows. Strengths include the open-source toolkit, integration of MLIPs with evolutionary search and MD validation, and demonstration on real databases. However, the overall significance hinges on demonstrating that MLIP-guided structures are not artifacts and remain stable under higher-accuracy methods.
major comments (3)
- [Results on specific material remediation] The claims of successful remediation for Cs₂KInI₆ and KTaSe₃ (abstract and results) provide no quantitative metrics such as MLIP vs. DFT energy differences, phonon frequency agreement, or false-positive rates for the final structures. This is load-bearing because MLIP inaccuracies in ionic or van der Waals interactions could produce spurious minima that appear stable only under the surrogate potential.
- [Methods and results on genetic algorithm performance] The assertion that the energy-guided genetic algorithm 'vastly outperforms' soft-mode following lacks any benchmarks, success rates, computational cost comparisons, or validation protocols on a test set (methods and results sections). Without these, the central performance claim cannot be assessed.
- [Application to Alexandria database] In the Alexandria database application, the identification of 'thousands of abandoned, high-symmetry stoichiometries' and 'dynamically stable low-symmetry candidates' is stated without specific examples, energy-above-hull values, or DFT cross-checks on the output structures. This undermines the broader applicability demonstration.
minor comments (2)
- [Abstract] The abstract would benefit from naming the specific foundational MLIPs employed and their training data sources to allow readers to assess transferability.
- [Methods on MD validation] Clarify in the methods whether the MD workflow includes anharmonic effects or merely checks retention of the 0 K structure, and ensure consistent terminology for 'dynamically stable' across sections.
Simulated Author's Rebuttal
We thank the referee for their thorough review and valuable feedback on our manuscript. We address each of the major comments below and outline the revisions we will make to strengthen the paper.
read point-by-point responses
-
Referee: [Results on specific material remediation] The claims of successful remediation for Cs₂KInI₆ and KTaSe₃ (abstract and results) provide no quantitative metrics such as MLIP vs. DFT energy differences, phonon frequency agreement, or false-positive rates for the final structures. This is load-bearing because MLIP inaccuracies in ionic or van der Waals interactions could produce spurious minima that appear stable only under the surrogate potential.
Authors: We agree that additional quantitative validation is essential to substantiate the remediation claims. In the revised manuscript, we will include a new table or section detailing the MLIP-predicted energies compared to DFT calculations for the final structures of Cs₂KInI₆ and KTaSe₃. We will also report phonon frequency comparisons for these structures. Regarding false-positive rates, we will discuss our multi-stage validation protocol, including finite-temperature MD simulations, and acknowledge the limitations of MLIPs in capturing certain interactions. We will perform DFT cross-validations on the key remediated structures to address concerns about spurious minima. revision: yes
-
Referee: [Methods and results on genetic algorithm performance] The assertion that the energy-guided genetic algorithm 'vastly outperforms' soft-mode following lacks any benchmarks, success rates, computational cost comparisons, or validation protocols on a test set (methods and results sections). Without these, the central performance claim cannot be assessed.
Authors: We acknowledge that the performance claim requires more rigorous benchmarking. We will add a dedicated subsection in the Methods and Results sections that includes benchmarks on a curated test set of materials with known instabilities. This will report success rates in finding stable polymorphs, computational costs (e.g., number of energy evaluations), and direct comparisons to soft-mode following methods. The test set will consist of small-to-medium sized systems where DFT is feasible for validation. This will allow readers to quantitatively assess the advantages of the energy-guided GA. revision: yes
-
Referee: [Application to Alexandria database] In the Alexandria database application, the identification of 'thousands of abandoned, high-symmetry stoichiometries' and 'dynamically stable low-symmetry candidates' is stated without specific examples, energy-above-hull values, or DFT cross-checks on the output structures. This undermines the broader applicability demonstration.
Authors: We agree that providing concrete examples and validation metrics would enhance the demonstration of broader applicability. In the revised manuscript, we will include specific examples of the identified low-symmetry candidates from the Alexandria database sample, along with their MLIP-computed energy-above-hull values. Additionally, we will perform and report DFT calculations for a subset of these candidates to provide cross-checks. This will be presented in a new figure or table in the Results section, illustrating the remediation process for these cases. revision: yes
Circularity Check
No circularity in VibroML toolkit description or claims
full rationale
The manuscript describes an engineering toolkit (VibroML) that combines existing MLIPs with a genetic algorithm for structural remediation, MD validation, and coupling to the authors' prior ProtoCSP engine. No equations, fitted parameters, or predictions are presented that reduce to inputs by construction. Claims of outperforming soft-mode following rest on reported applications to specific compounds and the Alexandria database rather than any self-referential derivation. Self-reference to ProtoCSP is acknowledged but is not load-bearing for any mathematical result; the toolkit's outputs are framed as externally validated via MD and phonon checks. This is a standard methods contribution with no detectable circular steps in its derivation chain.
Axiom & Free-Parameter Ledger
axioms (1)
- domain assumption Machine-learned interatomic potentials provide a sufficiently accurate representation of the potential energy surface for the crystalline materials under study.
Reference graph
Works this paper leans on
-
[1]
Shahzad, K., Mardare, A. I. & Hassel, A. W. Accelerating materials discovery: Combinatorial synthesis, high-throughput characterization, and computational advances.Science and Technology of Advanced Materials: Methods4, 2292486 (2024). 26
2024
-
[2]
L., Takeuchi, I
Joress, H., Green, M. L., Takeuchi, I. & Hattrick-Simpers, J. R. Applications of High Throughput (Combinatorial) Methodologies to Electronic, Magnetic, Structural, and Energy- Related Materials. InEncyclopedia of Materials: Metals and Alloys, 353–371 (Elsevier, 2022)
2022
-
[3]
Chen, C.et al.Accelerating Computational Materials Discovery with Machine Learning and Cloud High-Performance Computing: From Large-Scale Screening to Experimental Validation.Journal of the American Chemical Society146, 20009–20018 (2024)
2024
-
[4]
Bartel, C. J. Review of computational approaches to predict the thermodynamic stability of inorganic solids.Journal of Materials Science57, 10475–10498 (2022)
2022
-
[5]
Rudin, S. P. Generalization of soft phonon modes.Physical Review B97, 134114 (2018)
2018
-
[6]
Xiao, E. & Tadano, T. High-throughput computational screening of Heusler compounds with phonon considerations for enhanced material discovery.arXiv preprint(2025). 2502.17946
-
[7]
D., Xia, Y
Griesemer, S. D., Xia, Y. & Wolverton, C. Accelerating the prediction of stable materials with machine learning.Nature Computational Science3, 934–945 (2023)
2023
-
[8]
& Mizukami, W
Shiota, T., Ishihara, K. & Mizukami, W. Universal neural network potentials as descriptors: Towards scalable chemical property prediction using quantum and classical computers. Digital Discovery3, 1714–1728 (2024)
2024
-
[9]
Wang, J., Huang, Y., Gao, Z. & Du, J. Lead-Free Antimony-Based Perovskite Nanocrystals as a Fluorescent Nanoprobe for the Turn-On Detection of Trace Water in Organic Solvents. ACS Applied Nano Materials7, 7958–7965 (2024)
2024
-
[10]
Batzner, S.et al.E(3)-equivariant graph neural networks for data-efficient and accurate interatomic potentials.Nature Communications13, 2453 (2022)
2022
-
[11]
P., Simm, G
Batatia, I., Kov´ acs, D. P., Simm, G. N. C., Ortner, C. & Cs´ anyi, G. MACE: Higher Order Equivariant Message Passing Neural Networks for Fast and Accurate Force Fields (2022)
2022
-
[12]
& Jung, Y
Choi, J., Nam, G., Choi, J. & Jung, Y. A Perspective on Foundation Models in Chemistry. JACS Au5, 1499–1518 (2025)
2025
-
[13]
Musaelian, A.et al.Learning local equivariant representations for large-scale atomistic dynamics.Nature Communications14, 579 (2023)
2023
- [14]
-
[15]
Wood, B. M.et al.UMA: A Family of Universal Models for Atoms.arXiv preprint(2025). 2506.23971
-
[16]
S., Ghorbani, K., Hatam-Lee, S
Arabha, S., Aghbolagh, Z. S., Ghorbani, K., Hatam-Lee, S. M. & Rajabpour, A. Recent advances in lattice thermal conductivity calculation using machine-learning interatomic potentials.Journal of Applied Physics130, 210903 (2021)
2021
-
[17]
& Marques, M
Loew, A., Sun, D., Wang, H.-C., Botti, S. & Marques, M. A. L. Universal machine learning interatomic potentials are ready for phonons.npj Computational Materials11, 178 (2025)
2025
-
[18]
& Cao, B
Guo, L., Liu, Y., Yang, L. & Cao, B. Lattice dynamics modeling of thermal transport in solids using machine-learned atomic cluster expansion potentials: A tutorial.Journal of Applied Physics137, 081101 (2025). 27
2025
- [19]
-
[20]
& Cheng, Y
Han, B. & Cheng, Y. Benchmarking universal machine learning interatomic potentials for rapid analysis of inelastic neutron scattering data.Machine Learning: Science and Technology6, 030504 (2025)
2025
-
[21]
Radova, M., Stark, W. G., Allen, C. S., Maurer, R. J. & Bart´ ok, A. P. Fine-tuning foundation models of materials interatomic potentials with frozen transfer learning.arXiv preprint(2025).2502.15582
- [22]
-
[23]
Parackal, A. S., Trybel, F., Faber, F. A. & Armiento, R. Screening 39 billion protostructures for materials discovery.arXiv preprint(2026).2601.21393
- [24]
-
[25]
Han, X.-Q., Guo, P.-J., Gao, Z.-F. & Lu, Z.-Y. PhononBench:A Large-Scale Phonon- Based Benchmark for Dynamical Stability in Crystal Generation.arXiv preprint(2025). 2512.21227
-
[26]
& Tanaka, I
Togo, A. & Tanaka, I. Evolution of crystal structures in metallic elements.Physical Review B87, 184104 (2013)
2013
-
[27]
P., Simm, G., Ortner, C
Batatia, I., Kov´ acs, D. P., Simm, G., Ortner, C. & Cs´ anyi, G. MACE foundations: Pre-trained foundation models for atomistic materials chemistry. https://github.com/ ACEsuit/mace-foundations(2024). Accessed: 2025-02-10
2024
-
[28]
& Rabe, K
Qi, Y. & Rabe, K. M. Competing Phases of HfO 2 from Unstable Flat Phonon Bands of an Unconventional High-Symmetry Structure.Physical Review Letters135, 046101 (2025)
2025
-
[29]
Rahim, W.et al.Polymorph exploration of bismuth stannate using first-principles phonon mode mapping.Chemical Science11, 7904–7909 (2020)
2020
-
[30]
A., Bentria, B., Dahame, T
Fadla, M. A., Bentria, B., Dahame, T. & Benghia, A. First-principles investigation on the stability and material properties of all-inorganic cesium lead iodide perovskites CsPbI3 polymorphs.Physica B: Condensed Matter585, 412118 (2020)
2020
-
[31]
& Navrotsky, A
Wang, B., Novendra, N. & Navrotsky, A. Energetics, Structures, and Phase Transitions of Cubic and Orthorhombic Cesium Lead Iodide (CsPbI 3 ) Polymorphs.Journal of the American Chemical Society141, 14501–14504 (2019)
2019
-
[32]
Lv, S.et al.Physical origin of hafnium-based ferroelectricity.Computational Materials Today4, 100010 (2024)
2024
-
[33]
Pallikara, I., Kayastha, P., Skelton, J. M. & Whalley, L. D. The physical significance of imaginary phonon modes in crystals.Electronic Structure4, 033002 (2022)
2022
- [34]
-
[35]
Bakhsh, M., Trinquet, V., Gouvˆ ea, R. A., Rignanese, G.-M. & Ponc´ e, S. From Symmetry to Stability: Structural and Electronic Transformation in Cs$ 2$KInI$ 6$.arXiv preprint (2026).2602.06893. 28
-
[36]
& Yin, J
Bian, T., Zhu, W., Lei, Q. & Yin, J. Machine Learning Potentials for Inorganic and Hybrid Lead Halide Perovskites: From Phase Stability to Defects and Interfaces.ACS Applied Materials & Interfacesacsami.5c24460 (2026)
2026
-
[37]
J.et al.Dynamic Local Structure in Caesium Lead Iodide: Spatial Correlation and Transient Domains.Small20, 2303565 (2024)
Baldwin, W. J.et al.Dynamic Local Structure in Caesium Lead Iodide: Spatial Correlation and Transient Domains.Small20, 2303565 (2024)
2024
-
[38]
Braeckevelt, T.et al.Accurately Determining the Phase Transition Temperature of CsPbI 3 via Random-Phase Approximation Calculations and Phase-Transferable Machine Learning Potentials.Chemistry of Materials34, 8561–8576 (2022)
2022
-
[39]
& Sleight, A
Shannon, R., Bierlein, J., Gillson, J., Jones, G. & Sleight, A. Polymorphism in Bi2Sn2O7. Journal of Physics and Chemistry of Solids41, 117–122 (1980)
1980
-
[40]
W.et al.An Exhaustive Symmetry Approach to Structure Determination: Phase Transitions in Bi 2 Sn2 O7.Journal of the American Chemical Society138, 8031–8042 (2016)
Lewis, J. W.et al.An Exhaustive Symmetry Approach to Structure Determination: Phase Transitions in Bi 2 Sn2 O7.Journal of the American Chemical Society138, 8031–8042 (2016)
2016
-
[41]
Z.et al.A Comprehensive Assessment and Benchmark Study of Large Atomistic Foundation Models for Phonons.Advanced Intelligent Discoverye202500075 (2025)
Anam, M. Z.et al.A Comprehensive Assessment and Benchmark Study of Large Atomistic Foundation Models for Phonons.Advanced Intelligent Discoverye202500075 (2025)
2025
-
[42]
Babu, A., Gouvˆ ea, R. A., Vandergheynst, P. & Rignanese, G.-M. MEIDNet: Multimodal generative AI framework for inverse materials design.arXiv preprint(2026).2601.22009
-
[43]
Shannon, R. D. Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides.Acta Crystallographica Section A32, 751–767 (1976)
1976
-
[44]
npj Computational Materials11, 9 (2025)
Deng, B.et al.Systematic softening in universal machine learning interatomic potentials. npj Computational Materials11, 9 (2025)
2025
-
[45]
https://github.com/ACEsuit/mace-foundations
GitHub - ACEsuit/mace-foundations: MACE foundation models (MP, OMAT, Matpes). https://github.com/ACEsuit/mace-foundations
- [46]
-
[47]
Hjorth Larsen, A.et al.The atomic simulation environment - A Python library for working with atoms.Journal of Physics Condensed Matter29, 273002 (2017)
2017
-
[48]
PHON: A program to calculate phonons using the small displacement method
Alf` e, D. PHON: A program to calculate phonons using the small displacement method. Computer Physics Communications180, 2622–2633 (2009)
2009
-
[49]
First-principles Phonon Calculations with Phonopy and Phono3py.Journal of the Physical Society of Japan92, 012001 (2023)
Togo, A. First-principles Phonon Calculations with Phonopy and Phono3py.Journal of the Physical Society of Japan92, 012001 (2023)
2023
- [50]
-
[51]
Abraham, N. L. & Probert, M. I. J. Improved real-space genetic algorithm for crystal structure and polymorph prediction.Physical Review B77, 134117 (2008)
2008
-
[52]
Curtis, F.et al.GAtor: A First-Principles Genetic Algorithm for Molecular Crystal Structure Prediction.Journal of Chemical Theory and Computation14, 2246–2264 (2018)
2018
-
[53]
S., Wei, L., Hu, M
Omee, S. S., Wei, L., Hu, M. & Hu, J. Crystal structure prediction using neural network potential and age-fitness Pareto genetic algorithm.Journal of materials Informatics4 (2024). 29
2024
-
[54]
Piechota, J.et al.Ab Initio Molecular Dynamics Insight to Structural Phase Transition and Thermal Decomposition of InN.International Journal of Molecular Sciences25, 8281 (2024)
2024
-
[55]
& Ozgen, S
Kazanc, S., Ahmet Celik, F. & Ozgen, S. The investigation of solid–solid phase trans- formation at CuAlNi alloy using molecular dynamics simulation.Journal of Physics and Chemistry of Solids74, 1836–1841 (2013)
2013
-
[56]
P.et al.Python Materials Genomics (pymatgen): A robust, open-source python library for materials analysis.Computational Materials Science68, 314–319 (2013)
Ong, S. P.et al.Python Materials Genomics (pymatgen): A robust, open-source python library for materials analysis.Computational Materials Science68, 314–319 (2013)
2013
-
[57]
Hart, G. L. W. & Forcade, R. W. Algorithm for generating derivative structures.Physical Review B77, 224115 (2008)
2008
-
[58]
S., Hart, G
Morgan, W. S., Hart, G. L. & Forcade, R. W. Generating derivative superstructures for systems with high configurational freedom.Computational Materials Science136, 144–149 (2017)
2017
- [59]
-
[60]
LeMat-BulkUnique dataset (2023)
LeMaterial. LeMat-BulkUnique dataset (2023)
2023
-
[61]
& Furthm¨ uller, J
Kresse, G. & Furthm¨ uller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set.Physical Review B - Condensed Matter and Materials Physics 54, 11169–11186 (1996)
1996
-
[62]
P., Burke, K
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys Rev Lett77, 3865 (1996)
1996
-
[63]
& Joubert, D
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method.Physical Review B59, 1758–1775 (1999)
1999
-
[64]
[object Object] (2024)
Ganose, A.et al.Materialsproject/atomate2: V0.0.13. [object Object] (2024)
2024
-
[65]
K.et al.Accelerated data-driven materials science with the Materials Project
Horton, M. K.et al.Accelerated data-driven materials science with the Materials Project. Nature Materials24, 1522–1532 (2025)
2025
-
[66]
Kuhn, H. W. The Hungarian method for the assignment problem.Naval Research Logistics Quarterly2, 83–97 (1955). Acknowledgements Computational resources have been provided by the Consortium des ´Equipements de Calcul Intensif (C´ECI), funded by the Fonds de la Recherche Scientifique de Belgique (F.R.S.-FNRS) under Grant No. 2.5020.11 and by the Walloon Reg...
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.