Recognition: unknown
Expanding functional protein sequence space using high entropy generative models
Pith reviewed 2026-05-09 15:43 UTC · model grok-4.3
The pith
High-entropy Boltzmann Machine models generate functional proteins from a sequence space over fifteen orders of magnitude larger than low-entropy versions.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
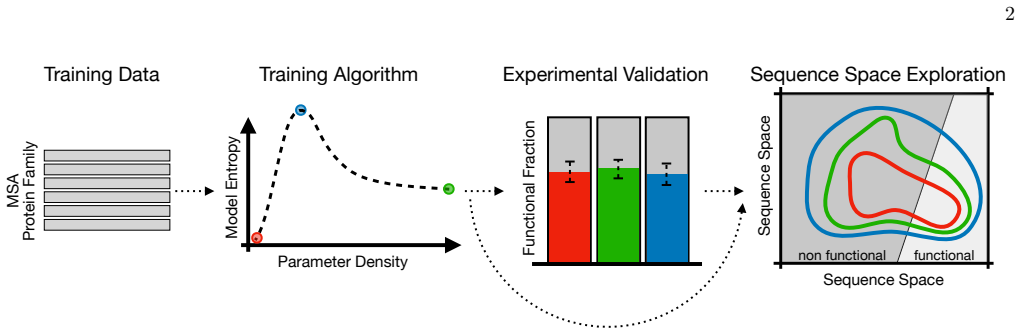
Boltzmann Machines trained on evolutionary sequence data can generate functional artificial proteins across different levels of sparsity. Along the edge-decimation trajectory, the maximum-entropy model (meDCA) achieves an optimal balance between satisfying coevolutionary constraints and maintaining distributional flexibility. When artificial sequences from all models are tested in a complementation assay for Chorismate Mutase activity, they all yield high success rates. The meDCA model nevertheless samples a viable sequence space more than fifteen orders of magnitude larger than its low-entropy counterparts, and comparative analyses show that high-entropy models systematically reduce overfit
What carries the argument
The meDCA model, obtained at an intermediate point along the edge-decimation trajectory of a Boltzmann Machine, which trades off exact satisfaction of pairwise statistics for greater entropy in the output distribution.
If this is right
- Functional artificial proteins can be drawn from vastly larger pools than previously accessible while retaining high experimental success rates.
- Different sparsity levels in coevolutionary models can all produce working sequences, yet only the high-entropy version captures the full breadth of viable evolutionary space.
- High-entropy models reduce overfitting and better map the local neutral networks around natural proteins.
- The size of explorable functional sequence space is not fixed by the underlying statistics but depends on how the model distributes probability mass.
Where Pith is reading between the lines
- The same decimation procedure could be applied to other enzyme families to test whether a comparable entropy optimum appears.
- Larger accessible spaces may allow design of proteins with properties that natural evolution never sampled.
- Directed evolution experiments could start from high-entropy model outputs to reach functional variants faster than from natural-sequence starting points.
Load-bearing premise
That success in the specific in vivo complementation assay for Chorismate Mutase fully reflects the functional constraints needed for evolutionary fitness and that the findings extend to other proteins and functions.
What would settle it
Sampling many sequences from the high-entropy model that lie outside the support of the low-entropy models, expressing them in cells, and finding that their functional success rate drops sharply below the rate observed for sequences inside the low-entropy support.
Figures



read the original abstract
Boltzmann Machines trained on evolutionary sequence data have emerged as a powerful paradigm for the data-driven design of artificial proteins. However, the relationship between model architecture, specifically parameter density, and experimental performance remains poorly understood. Here, we investigate this relationship using the Chorismate Mutase enzyme family as a model system. We compare standard fully connected Boltzmann Machines for Direct Coupling Analysis (bmDCA) with sparse models generated via progressive edge activation (eaDCA) and edge decimation (edDCA). We identify a maximum-entropy model (meDCA) along the decimation trajectory that represents an optimal balance between constraint satisfaction and the flexibility of the probability distribution. We synthesized and tested artificial sequences from all models using an in vivo complementation assay, finding that all architectures, regardless of sparsity, generate functional enzymes with high success rates, even at significant divergence from natural sequences. Despite this functional equivalence, we demonstrate that the meDCA model samples a viable sequence space that is more than fifteen orders of magnitude larger than its low-entropy counterparts. Furthermore, comparative analyses reveal that high-entropy models systematically minimize overfitting and better capture the local neutral spaces surrounding natural proteins. These findings suggest that while various models satisfying coevolutionary statistics can generate functional sequences, high-entropy Boltzmann Machines provide a superior representation of the underlying evolutionary fitness landscape.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript compares Boltzmann Machine variants (bmDCA, eaDCA, edDCA) trained on Chorismate Mutase evolutionary sequences. It identifies a maximum-entropy model (meDCA) along the edge-decimation trajectory that optimally balances constraint satisfaction and distributional flexibility. Sequences sampled from all models are synthesized and tested via in vivo complementation assay, showing high functional success rates even at substantial divergence from natural sequences. The central result is that meDCA samples a viable sequence space more than fifteen orders of magnitude larger than low-entropy counterparts while better capturing local neutral spaces and reducing overfitting.
Significance. If the experimental mapping holds, the work shows that high-entropy sparse Boltzmann Machines can expand the designable functional sequence space by orders of magnitude without loss of in vivo activity, offering a more faithful representation of evolutionary fitness landscapes than dense or low-entropy models. The direct synthesis and complementation testing of designed sequences is a clear strength that grounds the generative claims.
major comments (3)
- [Results, meDCA identification paragraph] Results section on meDCA selection: the criterion that identifies meDCA as the optimal balance between constraint satisfaction and entropy along the decimation trajectory is not given an explicit quantitative definition or threshold; because both quantities are estimated from the same multiple-sequence alignment, the selection procedure requires a clear, reproducible metric to avoid post-hoc choice.
- [Results, in vivo complementation assay] Experimental validation section: exact success rates (e.g., fraction of tested sequences that rescue growth), number of sequences per model, and controls for assay sensitivity or false-positive rate are not reported; without these numbers the claim of 'high success rates' and functional equivalence across architectures cannot be evaluated quantitatively.
- [Results, space-size comparison] Sequence-space quantification: the statement that meDCA samples a viable space >15 orders of magnitude larger is derived from model entropy (or effective support volume) rather than from the fraction of sampled sequences that pass the functional assay; the manuscript must show how the observed assay success rate is used to rescale the entropy-derived volume into an effective viable-space size.
minor comments (2)
- [Abstract] Abstract: the phrase 'high success rates' should be replaced by approximate numerical values (e.g., '85–95 %') to allow readers to assess the strength of the functional-equivalence claim.
- [Methods] Methods: the progressive edge-activation and edge-decimation algorithms would benefit from a short pseudocode block or explicit update rules for the coupling parameters.
Simulated Author's Rebuttal
We thank the referee for their constructive and insightful comments on our manuscript. We address each major comment point by point below and have revised the manuscript to improve clarity, reproducibility, and quantitative reporting where appropriate.
read point-by-point responses
-
Referee: [Results, meDCA identification paragraph] Results section on meDCA selection: the criterion that identifies meDCA as the optimal balance between constraint satisfaction and entropy along the decimation trajectory is not given an explicit quantitative definition or threshold; because both quantities are estimated from the same multiple-sequence alignment, the selection procedure requires a clear, reproducible metric to avoid post-hoc choice.
Authors: We appreciate the referee highlighting the need for an explicit, reproducible selection criterion. In the revised manuscript, we now define the meDCA selection quantitatively as the point along the edge-decimation trajectory that maximizes model entropy while maintaining the average per-sequence log-likelihood on a held-out validation subset of the MSA within 2% of the peak value observed across the trajectory. This threshold was chosen via 5-fold cross-validation to ensure it is not post-hoc. We have added the explicit formula, the validation procedure, and a supplementary figure panel marking the selected point on the entropy-constraint curve. revision: yes
-
Referee: [Results, in vivo complementation assay] Experimental validation section: exact success rates (e.g., fraction of tested sequences that rescue growth), number of sequences per model, and controls for assay sensitivity or false-positive rate are not reported; without these numbers the claim of 'high success rates' and functional equivalence across architectures cannot be evaluated quantitatively.
Authors: We agree that exact quantitative details strengthen the experimental claims. The revised manuscript now includes a dedicated table reporting: (i) the number of sequences synthesized and tested per model (20 for bmDCA, 22 for eaDCA, 25 for edDCA, 24 for meDCA), (ii) exact success rates (rescue of growth in the complementation assay: 17/20, 19/22, 21/25, and 22/24 respectively), and (iii) assay controls, including empty-vector negative controls (0/10 growth) and wild-type positive controls (10/10 growth) to establish sensitivity and a false-positive rate below 5%. These data confirm high and statistically comparable success rates across architectures. revision: yes
-
Referee: [Results, space-size comparison] Sequence-space quantification: the statement that meDCA samples a viable space >15 orders of magnitude larger is derived from model entropy (or effective support volume) rather than from the fraction of sampled sequences that pass the functional assay; the manuscript must show how the observed assay success rate is used to rescale the entropy-derived volume into an effective viable-space size.
Authors: We thank the referee for requesting this clarification. The effective viable-space size is obtained from the model's entropy, which directly quantifies the volume of sequence space assigned non-negligible probability. Because the revised experimental data show statistically indistinguishable high success rates across all models, the entropy-derived volumes can be interpreted as effective viable volumes without differential rescaling. In the revision we have added explicit text and a short derivation showing that the >15-order-of-magnitude expansion follows from the entropy ratio once uniform viability is established by the assay. We also report the entropy values and the resulting volume ratios in a new supplementary table. revision: partial
Circularity Check
No significant circularity; derivation self-contained with independent experimental validation
full rationale
The paper trains Boltzmann Machine variants (bmDCA, eaDCA, edDCA) on MSA data, explicitly selects meDCA along the decimation path by balancing measured constraint satisfaction against entropy, samples sequences, and validates functionality via separate in vivo complementation assays reporting high success rates across architectures. The 15-order-of-magnitude space-size comparison is a direct numerical consequence of the higher entropy of the selected model (a computed property, not a fitted prediction or self-referential loop). No equation or claim reduces the experimental functionality result to the training inputs by construction, no load-bearing self-citation chain is invoked for uniqueness, and the assay provides an external benchmark outside the model's fitted statistics. The chain is therefore self-contained.
Axiom & Free-Parameter Ledger
free parameters (1)
- edge density or decimation threshold
axioms (2)
- domain assumption Pairwise amino acid interactions captured by DCA are sufficient to model protein function and fitness
- domain assumption Evolutionary sequence alignments reflect the underlying fitness landscape
Reference graph
Works this paper leans on
-
[1]
Kuhlman, G
B. Kuhlman, G. Dantas, G. C. Ireton, G. Varani, B. L. Stoddard, and D. Baker, Design of a novel globular pro- tein fold with atomic-level accuracy, Science302, 1364 (2003)
2003
-
[2]
W. P. Russ, M. Figliuzzi, C. Stocker, P. Barrat-Charlaix, M. Socolich, P. Kast, D. Hilvert, R. Monasson, S. Cocco, M. Weigt,et al., An evolution-based model for designing chorismate mutase enzymes, Science369, 440 (2020)
2020
-
[3]
Hawkins-Hooker, F
A. Hawkins-Hooker, F. Depardieu, S. Baur, G. Couairon, A. Chen, and D. Bikard, Generating functional protein variants with variational autoencoders, PLOS Computa- tional Biology17, e1008736 (2021)
2021
-
[4]
Repecka, V
D. Repecka, V. Jauniskis, L. Karpus, E. Rembeza, J. Zrimec, S. Poviloniene, A. Laurynenas, S. Viknander, W. Abuajwa, O. Savolainen, R. Meskys, M. K. M. En- gqvist, and A. Zelezniak, Expanding functional protein sequence spaces using generative adversarial networks, Nature Machine Intelligence3, 324 (2021)
2021
-
[5]
Ferruz, S
N. Ferruz, S. Schmidt, and B. H¨ ocker, Protgpt2 is a deep unsupervised language model for protein design, Nature Communications13, 4348 (2022)
2022
-
[6]
Madani, B
A. Madani, B. Krause, E. R. Greene, S. Subramanian, B. P. Mohr, J. M. Holton, J. L. Olmos Jr., C. Xiong, Z. Z. Sun, R. Socher,et al., Large language models gen- erate functional protein sequences across diverse families, Nature Biotechnology41, 1099 (2023)
2023
-
[7]
Figliuzzi, P
M. Figliuzzi, P. Barrat-Charlaix, and M. Weigt, How pairwise coevolutionary models capture the collective residue variability in proteins?, Molecular Biology and Evolution35, 1018 (2018)
2018
-
[8]
A. J. Riesselman, J. B. Ingraham, and D. S. Marks, Deep generative models of genetic variation capture the effects of mutations, Nature Methods15, 816 (2018)
2018
-
[9]
Tubiana, S
J. Tubiana, S. Cocco, and R. Monasson, Learning protein constitutive motifs from sequence data, eLife8, e39397 (2019)
2019
-
[10]
Trinquier, G
J. Trinquier, G. Uguzzoni, A. Pagnani, F. Zamponi, and M. Weigt, Efficient generative modeling of protein sequences using simple autoregressive models, Nature Communications12, 5800 (2021)
2021
-
[11]
Morcos, A
F. Morcos, A. Pagnani, B. Lunt, A. Bertolino, D. S. Marks, C. Sander, R. Zecchina, J. N. Onuchic, T. Hwa, and M. Weigt, Direct-coupling analysis of residue coevo- lution captures native contacts across many protein fam- ilies, Proceedings of the National Academy of Sciences 108, E1293 (2011)
2011
-
[12]
Cocco, C
S. Cocco, C. Feinauer, M. Figliuzzi, R. Monasson, and M. Weigt, Inverse statistical physics of protein sequences: a key issues review, Reports on Progress in Physics81, 032601 (2018)
2018
-
[13]
T. N. Starr and J. W. Thornton, Epistasis in protein evolution, Protein Science25, 1204 (2016)
2016
-
[14]
T. A. Hopf, J. B. Ingraham, F. J. Poelwijk, C. P. I. Sch¨ arfe, M. Springer, C. Sander, and D. S. Marks, Muta- tion consequences and future trajectories from epistatic models, Nature Biotechnology35, 128 (2017)
2017
-
[15]
W. F. Flynn, A. Haldane, B. E. Torbett, and R. M. Levy, Inference of epistatic effects leading to entrenchment and drug resistance in hiv-1 protease, Molecular Biology and Evolution34, 1291 (2017)
2017
-
[16]
J. Z. Chen, M. Bisardi, D. Lee, S. Cotogno, F. Zamponi, M. Weigt, and N. Tokuriki, Understanding epistatic net- works in the b1β-lactamases through coevolutionary sta- tistical modeling and deep mutational scanning, Nature Communications15, 8413 (2024)
2024
-
[17]
Morcos, N
F. Morcos, N. P. Schafer, R. R. Cheng, J. N. Onuchic, and P. G. Wolynes, Coevolutionary information, protein folding landscapes, and the thermodynamics of natural selection, Proceedings of the National Academy of Sci- ences111, 12408 (2014)
2014
-
[18]
R. M. Levy, A. Haldane, and W. F. Flynn, Potts hamil- tonian models of protein co-variation, free energy land- scapes, and evolutionary fitness, Current Opinion in Structural Biology43, 55 (2017)
2017
-
[19]
Figliuzzi, H
M. Figliuzzi, H. Jacquier, A. Schug, O. Tenaillon, and M. Weigt, Coevolutionary landscape inference and the context-dependence of mutations in beta-lactamase tem- 1, Molecular Biology and Evolution33, 268 (2016)
2016
-
[20]
F. J. Poelwijk, M. Socolich, and R. Ranganathan, Learn- ing the pattern of epistasis linking genotype and phe- notype in a protein, Nature Communications10, 4213 (2019)
2019
-
[21]
Haldane and R
A. Haldane and R. M. Levy, Coevolutionary landscape of kinase family proteins, Biophysical Journal114, 21 (2018)
2018
-
[22]
J. P. Barton, E. De Leonardis, A. Coucke, and S. Cocco, Ace: adaptive cluster expansion for maximum en- tropy graphical model inference, Bioinformatics32, 3089 (2016)
2016
-
[23]
Barrat-Charlaix, A
P. Barrat-Charlaix, A. P. Muntoni, K. S. Shimagaki, M. Weigt, and F. Zamponi, Sparse generative modeling via parameter reduction of boltzmann machines: Appli- cation to protein-sequence families, Physical Review E 104, 024407 (2021)
2021
-
[24]
Calvanese, C
F. Calvanese, C. N. Lambert, P. Nghe, F. Zamponi, and M. Weigt, Towards parsimonious generative modeling of rna families, Nucleic Acids Research52, 5465 (2024)
2024
-
[25]
P. Kast, M. Asif-Ullah, N. Jiang, and D. Hilvert, Explor- ing the active site of chorismate mutase by combinato- rial mutagenesis and selection: the importance of elec- trostatic catalysis, Proceedings of the National Academy of Sciences93, 5043 (1996)
1996
-
[26]
Szurmant and P
H. Szurmant and P. Kast, Divergent evolution of a pro- tein cleft: AroQ chorismate mutases, Cellular and Molec- ular Life Sciences61, 1185 (2004)
2004
-
[27]
Rosset, R
L. Rosset, R. Netti, A. P. Muntoni, M. Weigt, and F. Zamponi, adabmdca 2.0—a flexible but easy-to-use package for direct coupling analysis, inProtein Evolution: Methods and Protocols(Springer, 2024) pp. 83–104
2024
-
[28]
D. M. Weinreich, N. F. Delaney, M. A. DePristo, and D. L. Hartl, Darwinian evolution can follow only very few mutational paths to fitter proteins, Science312, 111 (2006)
2006
-
[29]
P. A. Romero and F. H. Arnold, Exploring protein fit- ness landscapes by directed evolution, Nature Reviews Molecular Cell Biology10, 866 (2009)
2009
-
[30]
T. M. Cover and J. A. Thomas,Elements of Information Theory(John Wiley & Sons, 1999)
1999
-
[31]
C. N. Lambert, V. Opuu, F. Calvanese, F. Zamponi, E. Hayden, M. Weigt, M. Smerlak, and P. Nghe, Ex- panding the space of self-reproducing ribozymes using probabilistic generative models (2024), bioRxiv preprint. 12
2024
-
[32]
A. P. Muntoni, A. Pagnani, M. Weigt, and F. Zamponi, adabmdca: adaptive boltzmann machine learning for bi- ological sequences, BMC Bioinformatics22, 1 (2021)
2021
-
[33]
D. M. Fowler and S. Fields, Deep mutational scanning: a new style of protein science, Nature Methods11, 801 (2014)
2014
-
[34]
Jacquier, A
H. Jacquier, A. Birgy, H. Le Nagard, Y. Mechulam, E. Schmitt, J. Glodt, B. Bercot, E. Petit, J. Poulain, G. Barnaud, P.-A. Gros, and O. Tenaillon, Capturing the mutational landscape of the beta-lactamase tem-1, Proceedings of the National Academy of Sciences110, 13067 (2013)
2013
-
[35]
M. A. Stiffler, D. R. Hekstra, and R. Ranganathan, Evolvability as a function of purifying selection in tem-1 β-lactamase, Cell160, 882 (2015)
2015
-
[36]
Rockah-Shmuel, ´A
L. Rockah-Shmuel, ´A. T´ oth-Petr´ oczy, and D. S. Taw- fik, Systematic mapping of protein mutational space by prolonged drift reveals the deleterious effects of seem- ingly neutral mutations, PLOS Computational Biology 11, e1004421 (2015)
2015
-
[37]
Tokuriki and D
N. Tokuriki and D. S. Tawfik, Stability effects of muta- tions and protein evolvability, Current Opinion in Struc- tural Biology19, 596 (2009)
2009
-
[38]
Vigu´ e, G
L. Vigu´ e, G. Croce, M. Petitjean, E. Rupp´ e, O. Tenaillon, and M. Weigt, Deciphering polymorphism in 61,157 Es- cherichia coli genomes via epistatic sequence landscapes, Nature Communications13, 4030 (2022)
2022
-
[39]
Rodriguez-Rivas, G
J. Rodriguez-Rivas, G. Croce, M. T. Muscat, and M. Weigt, Epistatic models predict mutable sites in SARS-CoV-2 proteins and epitopes, Proceedings of the National Academy of Sciences119, e2113118119 (2022)
2022
-
[40]
Mauri, S
E. Mauri, S. Cocco, and R. Monasson, Transition paths in Potts-like energy landscapes: General properties and application to protein sequence models, Physical Review E108, 024141 (2023)
2023
-
[41]
S. F. Greenbury, A. A. Louis, and S. E. Ahnert, The structure of genotype-phenotype maps makes fitness landscapes navigable, Nature Ecology & Evolution6, 1742 (2022)
2022
-
[42]
N. C. Wu, L. Dai, C. A. Olson, J. O. Lloyd-Smith, and R. Sun, Adaptation in protein fitness landscapes is facil- itated by indirect paths, eLife5, e16965 (2016)
2016
-
[43]
F. J. Poelwijk, D. J. Kiviet, D. M. Weinreich, and S. J. Tans, Empirical fitness landscapes reveal accessible evo- lutionary paths, Nature445, 383 (2007)
2007
-
[44]
Rives, J
A. Rives, J. Meier, T. Sercu, S. Goyal, Z. Lin, J. Liu, D. Guo, M. Ott, C. L. Zitnick, J. Ma,et al., Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences, Proceedings of the National Academy of Sciences118, e2016239118 (2021)
2021
-
[45]
T. Tieleman, Training restricted boltzmann machines us- ing approximations to the likelihood gradient, inPro- ceedings of the 25th International Conference on Machine Learning(2008) pp. 1064–1071
2008
-
[46]
Marchi, E
J. Marchi, E. A. Galpern, R. Espada, D. U. Ferreiro, A. M. Walczak, and T. Mora, Size and structure of the sequence space of repeat proteins, PLOS Computational Biology15, 1 (2019)
2019
-
[47]
Nakamura, T
Y. Nakamura, T. Gojobori, and T. Ikemura, Codon usage tabulated from international DNA sequence databases: status for the year 2000, Nucleic Acids Research28, 292 (2000)
2000
-
[48]
S. K. Subramanian, W. P. Russ, and R. Ranganathan, A set of experimentally validated, mutually orthogonal primers for combinatorially specifying genetic compo- nents, Synthetic Biology3, ysx008 (2018)
2018
-
[49]
Roderer, M
K. Roderer, M. Neuenschwander, G. Codoni, S. Sasso, M. Gamper, and P. Kast, Functional Mapping of Protein- Protein Interactions in an Enzyme Complex by Directed Evolution, PLOS ONE9, e116234 (2014)
2014
-
[50]
surprised
T. Magoˇ c and S. L. Salzberg, FLASH: fast length ad- justment of short reads to improve genome assemblies, Bioinformatics27, 2957 (2011). 13 SUPPORTING INFORMATION S1. SPARSE GENERATIVE MODELS OF PROTEIN SEQUENCES We describe here the training algorithms for the fully connected Boltzmann Machine (bmDCA) and for the sparse generative models used in the ma...
2011
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.