Recognition: unknown
AgenticPosesRanker: An Agentic AI Framework for Physically Grounded Ranking of Protein-Ligand Docking Poses
Pith reviewed 2026-05-09 15:31 UTC · model grok-4.3
The pith
Agentic AI framework matches Smina scoring function accuracy in ranking protein-ligand docking poses.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
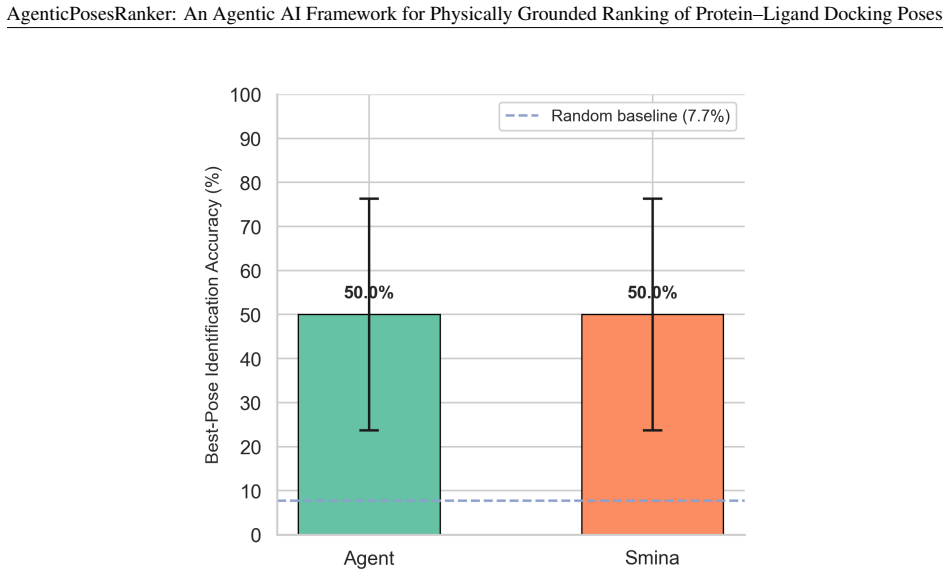
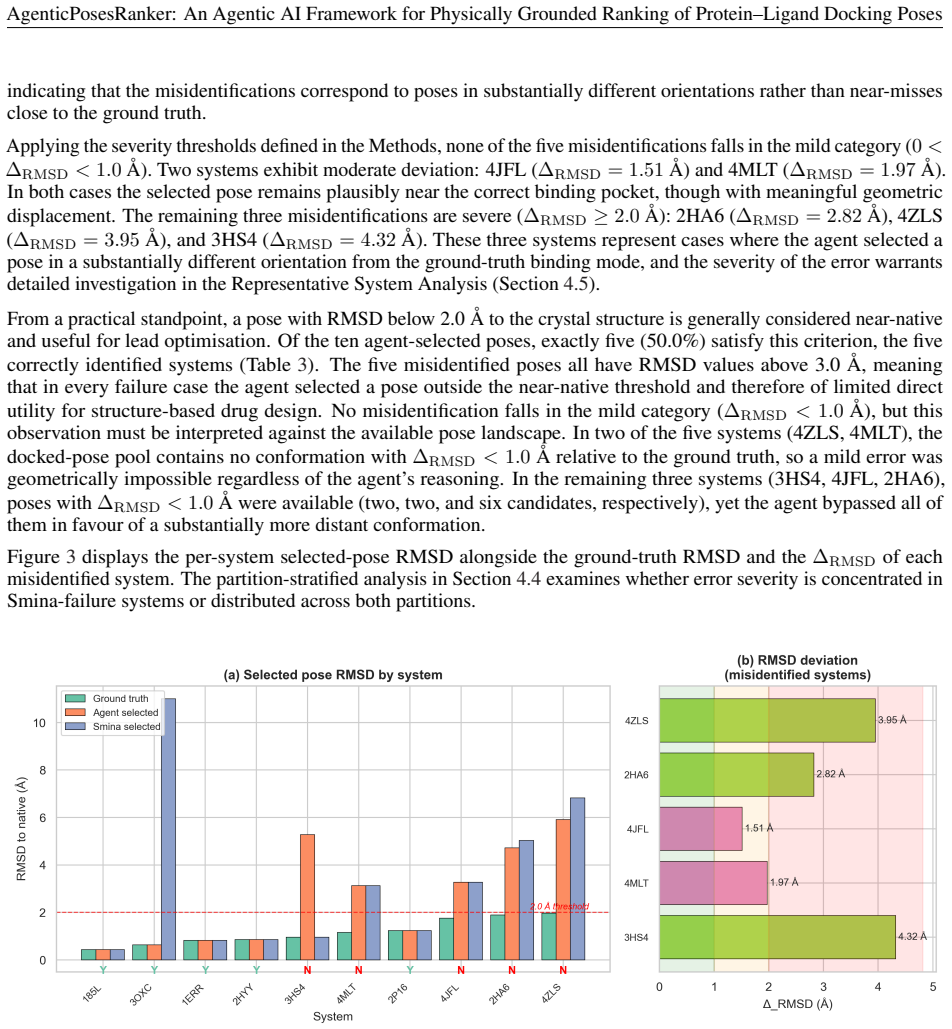
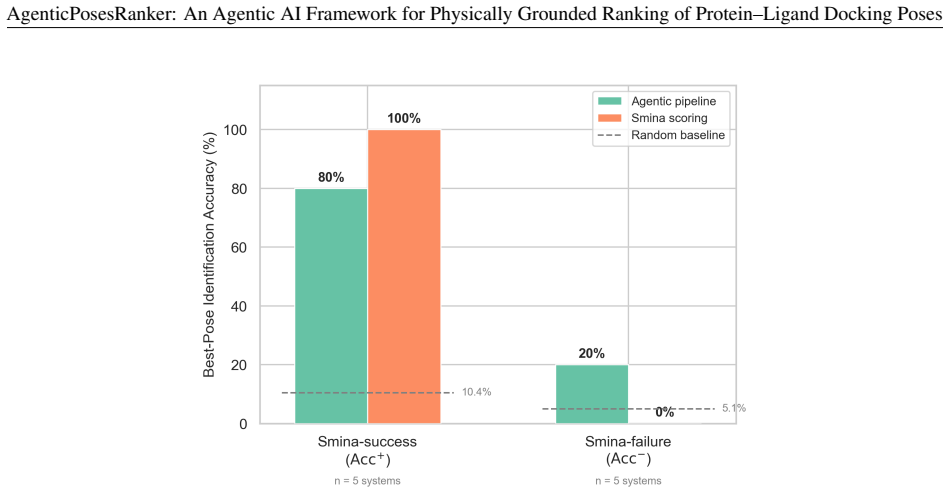
The agent achieved 50.0 percent best-pose accuracy on the balanced benchmark, matching the design-fixed Smina baseline of 50.0 percent while retaining 80 percent of Smina-success systems and recovering 20 percent of Smina-failure systems. Decision-attribution analysis showed high alignment between the agent's self-reported tool weights and objective metric separations of the selected pose, with median correlation of 0.83 across both correct and incorrect outcomes. These results position the framework as an interpretable curation layer for late-stage pose refinement rather than a net improvement over the reference scorer.
What carries the argument
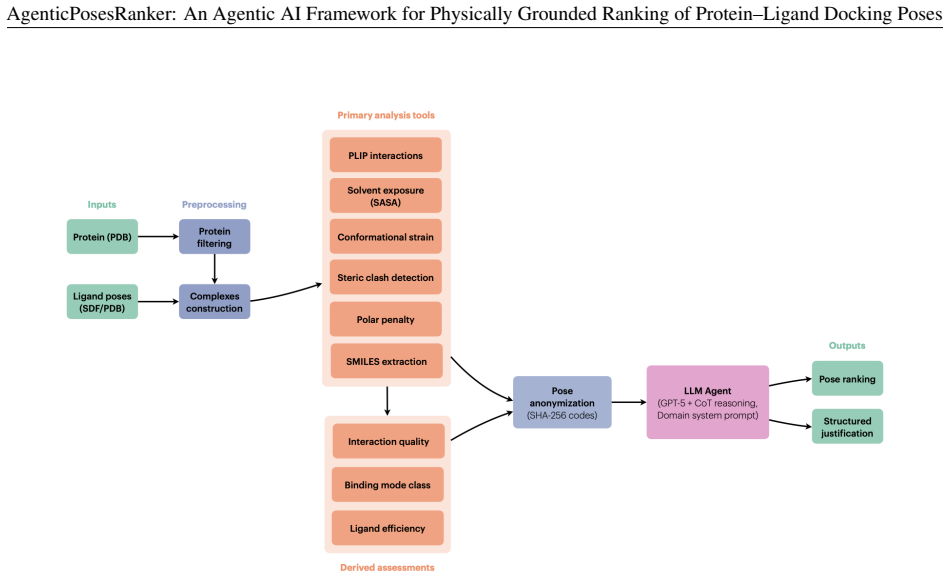
The chain-of-thought agent that applies the six deterministic tools in sequence (interaction fingerprinting, solvent-accessible burial, conformational strain, steric-clash detection, unsatisfied-polar-atom penalty, and chemical-identity extraction) to produce ranked evaluations of docking poses.
If this is right
- The agent offsets one regression with one recovery to maintain overall parity with the Smina baseline.
- High correlation between reported tool weights and metric separations localizes the performance limit to tool coverage rather than reasoning inconsistency.
- The approach supplies a template for evaluating agentic AI systems against objective ground truth in the natural sciences.
- The framework can serve as an interpretable post-processing layer for late-stage pose refinement in structure-based drug design.
Where Pith is reading between the lines
- Expanding the tool suite to include additional factors such as dynamic protein motions could raise the observed performance ceiling.
- The same agentic structure of deterministic tools plus language-model reasoning could be tested on related ranking problems such as binding-mode selection across multiple ligands.
- Integrating the curation layer upstream of improved base docking engines could produce additive gains in overall pose quality.
Load-bearing premise
The six chosen deterministic tools together capture all the main physicochemical factors that determine correct pose ranking.
What would settle it
A new benchmark set where the agent's accuracy falls significantly below 50 percent while Smina remains at 50 percent, or where tool attributions show low correlation with objective metrics, would indicate that the current tool suite does not fully cover the deciding factors.
Figures
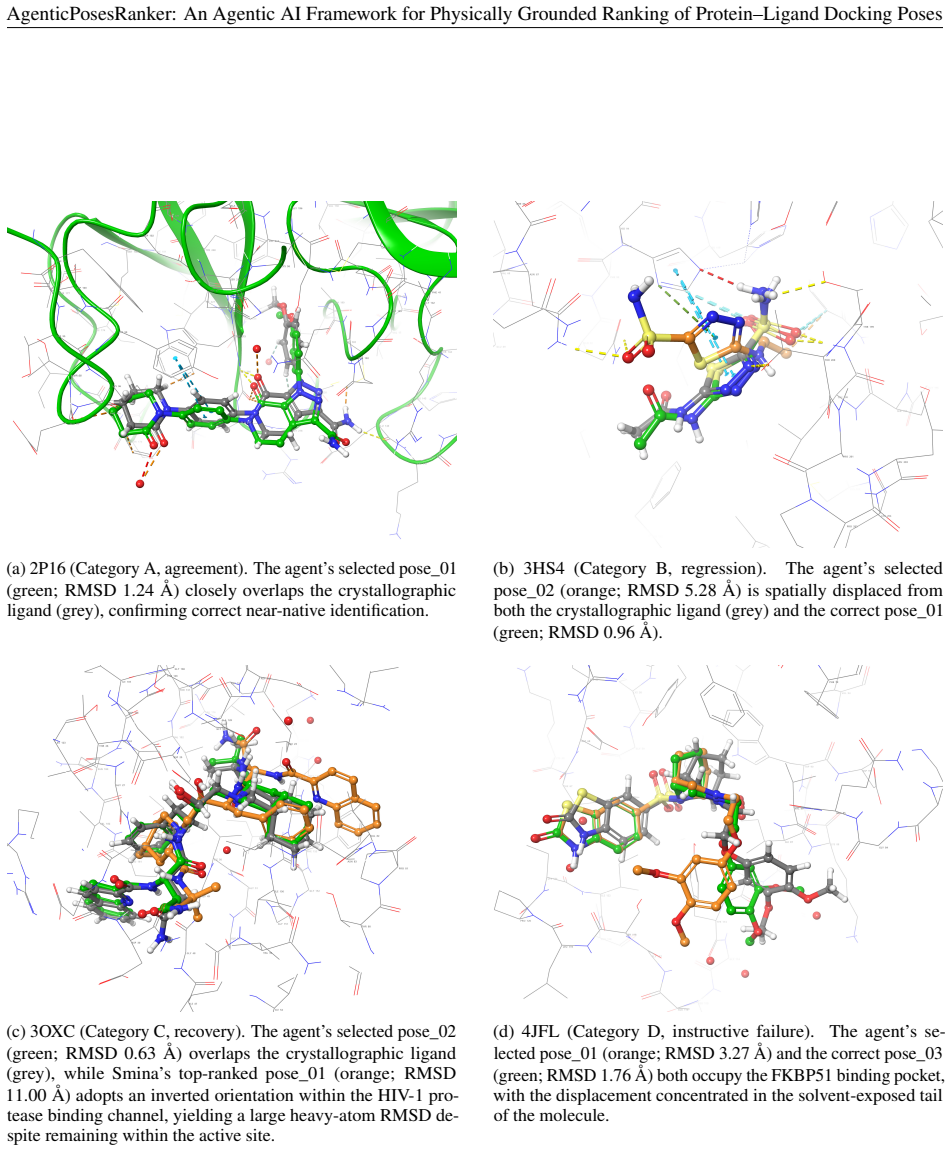





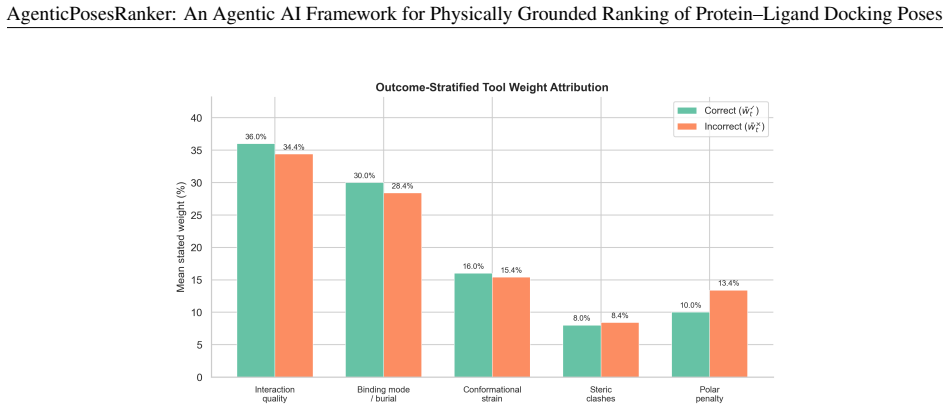
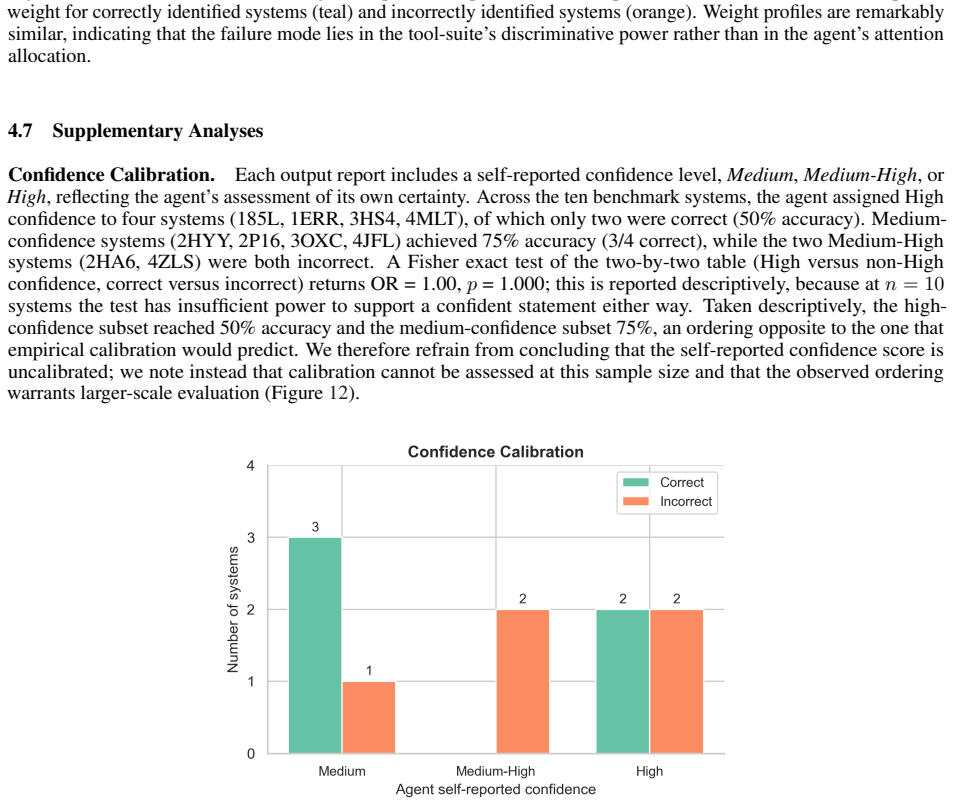


read the original abstract
Scoring functions remain the principal bottleneck in molecular docking: they routinely fail to rank near-native poses above decoys, and their composite single-score design obscures the physicochemical basis of each ranking error. We present AgenticPosesRanker, an agentic AI framework that combines six deterministic, physically grounded analysis tools (interaction fingerprinting, solvent-accessible burial, conformational strain, steric-clash detection, unsatisfied-polar-atom penalty, and chemical-identity extraction) with large-language-model (GPT-5) chain-of-thought reasoning to evaluate and rank docking poses. On a curated benchmark of ten protein-ligand systems (162 poses) balanced by construction between Smina scoring-function successes and failures, the agent achieved 50.0% best-pose accuracy, matching the design-fixed Smina baseline of 50.0% and significantly exceeding a 7.7% uniformly random baseline (p < 0.001, one-sided exact binomial test). The balanced-benchmark accuracy decomposes symmetrically: the agent retained 80% (4/5) of the Smina-success systems and recovered 20% (1/5) of the Smina-failure systems, so the aggregate 50% reflects one regression offset by one recovery rather than any net improvement over the Smina reference. Decision-attribution analysis showed high alignment between the agent's self-reported tool weights and objective metric separations of the selected pose (median \r{ho} = +0.83), consistent across correct and incorrect outcomes, localising the performance ceiling to tool-suite coverage rather than reasoning inconsistency. These results establish a methodological template for evaluating agentic AI against objective ground truth in the natural sciences and position the framework as an interpretable curation layer for late-stage pose refinement in structure-based drug design.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript introduces AgenticPosesRanker, an agentic AI framework that integrates six deterministic, physically grounded tools (interaction fingerprinting, solvent-accessible burial, conformational strain, steric-clash detection, unsatisfied-polar-atom penalty, and chemical-identity extraction) with GPT-5 chain-of-thought reasoning to evaluate and rank protein-ligand docking poses. On a curated benchmark of ten protein-ligand systems (162 poses) balanced by construction between Smina scoring successes and failures, the agent reports 50.0% best-pose accuracy, matching the Smina baseline and exceeding a uniform random baseline (p < 0.001, one-sided exact binomial test). The accuracy decomposes symmetrically as 80% retention of Smina successes and 20% recovery of Smina failures; decision-attribution analysis shows high alignment (median ρ = +0.83) between self-reported tool weights and objective metric separations. The paper claims these results establish a methodological template for evaluating agentic AI against objective ground truth in the natural sciences and position the framework as an interpretable curation layer for late-stage pose refinement in structure-based drug design.
Significance. If the framework's performance generalizes, it would offer a useful interpretable alternative to opaque composite scoring functions by decomposing pose rankings into explicit physicochemical factors, potentially aiding late-stage refinement in drug design. Strengths include the explicit symmetric decomposition of performance, the statistical comparison to both Smina and random baselines, and the correlation analysis linking LLM tool weights to objective metrics, all of which support interpretability and reproducibility. However, the absence of net improvement over Smina and the deliberately balanced small benchmark constrain the immediate practical significance and the strength of the methodological-template claim.
major comments (2)
- [Abstract] Abstract: The benchmark is deliberately balanced by construction (5 Smina-success and 5 Smina-failure systems), producing the reported 50% accuracy via symmetric offset (4/5 retained successes, 1/5 recovered failures) with zero net gain over the Smina reference. Real docking pose distributions are not pre-balanced 50/50, so the design does not test curation value under typical conditions and thereby weakens support for the claim that the results position the framework as an interpretable curation layer.
- [Abstract] Abstract: The assertion that the results 'establish a methodological template for evaluating agentic AI against objective ground truth' rests on only ten systems. With this small N, the median ρ = +0.83 alignment and its consistency across correct/incorrect outcomes are observed on too few decisions to support generalization to a template without additional validation on larger, unbalanced sets.
minor comments (1)
- [Abstract] Abstract: The notation 'median r{ho} = +0.83' is a LaTeX rendering error and should be corrected to 'median ρ = +0.83' (Spearman rank correlation).
Simulated Author's Rebuttal
Thank you for the constructive comments on our manuscript. We have carefully considered each point and provide point-by-point responses below. Where revisions are warranted, we indicate the changes to be made in the revised version.
read point-by-point responses
-
Referee: [Abstract] Abstract: The benchmark is deliberately balanced by construction (5 Smina-success and 5 Smina-failure systems), producing the reported 50% accuracy via symmetric offset (4/5 retained successes, 1/5 recovered failures) with zero net gain over the Smina reference. Real docking pose distributions are not pre-balanced 50/50, so the design does not test curation value under typical conditions and thereby weakens support for the claim that the results position the framework as an interpretable curation layer.
Authors: We agree with the observation that the benchmark was constructed to be balanced, resulting in no net improvement over the Smina baseline. This design was intentional to clearly demonstrate the agent's capacity to both retain correct rankings and recover from incorrect ones, providing insight into its interpretability through the symmetric decomposition. However, we acknowledge that this does not directly evaluate performance on the imbalanced distributions typical in real docking scenarios. To strengthen the manuscript, we will revise the abstract and add a dedicated limitations paragraph in the discussion section to explicitly state that future work will involve testing on larger, unbalanced benchmarks to better assess its value as a curation layer in practical settings. This revision will moderate the positioning claim accordingly. revision: partial
-
Referee: [Abstract] Abstract: The assertion that the results 'establish a methodological template for evaluating agentic AI against objective ground truth' rests on only ten systems. With this small N, the median ρ = +0.83 alignment and its consistency across correct/incorrect outcomes are observed on too few decisions to support generalization to a template without additional validation on larger, unbalanced sets.
Authors: The referee is correct that the current benchmark comprises only ten systems, limiting the strength of claims regarding a general methodological template. The high alignment in decision attribution (median ρ = +0.83) is encouraging but indeed based on a modest number of cases. We will revise the abstract to describe the work as providing an initial methodological template or proof-of-concept for such evaluations, rather than claiming it fully establishes one. Additionally, we will expand the discussion to include the small sample size as a limitation and outline plans for scaling the benchmark in subsequent studies. This addresses the concern about generalization. revision: yes
Circularity Check
No circularity: performance metrics rest on external ground truth and explicit benchmark design
full rationale
The paper reports observed accuracy (50%) on a benchmark explicitly balanced by construction between Smina successes and failures, with the agent's result decomposed transparently into 4/5 retained successes and 1/5 recovered failures. This is compared to an independent Smina baseline (also 50% by design) and a uniform random baseline (7.7%), with statistical testing against ground-truth correct poses. Tool-weight alignment (median ρ=+0.83) is computed as correlation to objective metric separations, not fitted or self-defined. No equations, parameters, or derivations reduce the reported outcomes to inputs by construction. No self-citations, uniqueness claims, or ansatzes appear as load-bearing elements. The framework's internal logic (tools + LLM reasoning) remains independent of the benchmark selection and accuracy numbers.
Axiom & Free-Parameter Ledger
axioms (1)
- domain assumption The six deterministic tools (interaction fingerprinting, solvent-accessible burial, conformational strain, steric-clash detection, unsatisfied-polar-atom penalty, and chemical-identity extraction) are collectively sufficient to evaluate and rank docking poses.
Reference graph
Works this paper leans on
-
[1]
Oleg Trott and Arthur J. Olson. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading.Journal of Computational Chemistry, 31(2):455–461, 2010
2010
-
[2]
A medicinal chemist’s guide to molecular interactions.Journal of Medicinal Chemistry, 53(14):5061–5084, 2010
Caterina Bissantz, Bernd Kuhn, and Martin Stahl. A medicinal chemist’s guide to molecular interactions.Journal of Medicinal Chemistry, 53(14):5061–5084, 2010
2010
-
[3]
Greenidge, Christian Kramer, Jean-Christophe Mozziconacci, and Woody Sherman
Paulette A. Greenidge, Christian Kramer, Jean-Christophe Mozziconacci, and Woody Sherman. Improving docking results via reranking of ensembles of ligand poses in multiple X-ray protein conformations with MM-GBSA. Journal of Chemical Information and Modeling, 54(10):2697–2717, 2014
2014
-
[4]
Warren, C
Gregory L. Warren, C. Webster Andrews, Anna-Maria Capelli, Brian Clarke, Judith LaLonde, Millard H. Lambert, Mika Lindvall, Neysa Nevins, Simon F. Semus, Stefan Senger, et al. A critical assessment of docking programs and scoring functions.Journal of Medicinal Chemistry, 49(20):5912–5931, 2006
2006
-
[5]
Baumgartner, and Carlos J
David Ryan Koes, Matthew P. Baumgartner, and Carlos J. Camacho. Lessons learned in empirical scoring with Smina from the CSAR 2011 benchmarking exercise.Journal of Chemical Information and Modeling, 53(8):1893–1904, 2013
2011
-
[6]
Najmanovich
Francis Gaudreault and Rafael J. Najmanovich. FlexAID: Revisiting docking on non-native-complex structures. Journal of Chemical Information and Modeling, 55(7):1323–1336, 2015
2015
-
[7]
Zhe Wang, Huiyong Sun, Xiaojun Yao, Dan Li, Lei Xu, Youyong Li, Sheng Tian, and Tingjun Hou. Comprehensive evaluation of ten docking programs on a diverse set of protein–ligand complexes: The prediction accuracy of sampling power and scoring power.Physical Chemistry Chemical Physics, 18(18):12964–12975, 2016
2016
-
[8]
Comparative assessment of scoring functions: The CASF-2016 update.Journal of Chemical Information and Modeling, 59(2):895–913, 2019
Minyi Su, Qifan Yang, Yu Du, Guoqin Feng, Zhihai Liu, Yan Li, and Renxiao Wang. Comparative assessment of scoring functions: The CASF-2016 update.Journal of Chemical Information and Modeling, 59(2):895–913, 2019
2016
-
[9]
Hartshorn, Marcel L
Michael J. Hartshorn, Marcel L. Verdonk, Gianni Chessari, Suzanne C. Brewerton, Wijnand T. M. Mooij, Paul N. Mortenson, and Christopher W. Murray. Diverse, high-quality test set for the validation of protein–ligand docking performance.Journal of Medicinal Chemistry, 50(4):726–741, 2007
2007
-
[10]
Rocco Meli and Philip C. Biggin. spyrmsd: symmetry-corrected RMSD calculations in Python.Journal of Cheminformatics, 12(1):49, 2020
2020
-
[11]
Smith, Ying Yang, John J
Shuo Gu, Matthew S. Smith, Ying Yang, John J. Irwin, and Brian K. Shoichet. Ligand strain energy in large library docking.Journal of Chemical Information and Modeling, 61(9):4331–4341, 2021
2021
-
[12]
Morris, and Charlotte M
Martin Buttenschoen, Garrett M. Morris, and Charlotte M. Deane. PoseBusters: AI-based docking methods fail to generate physically valid poses or generalise to novel sequences.Chemical Science, 15(9):3130–3139, 2024
2024
-
[13]
Knowledge-based scoring function to predict protein– ligand interactions.Journal of Molecular Biology, 295(2):337–356, 2000
Holger Gohlke, Manfred Hendlich, and Gerhard Klebe. Knowledge-based scoring function to predict protein– ligand interactions.Journal of Molecular Biology, 295(2):337–356, 2000
2000
-
[14]
Ingo Muegge and Yvonne C. Martin. A general and fast scoring function for protein–ligand interactions: A simplified potential approach.Journal of Medicinal Chemistry, 42(5):791–804, 1999
1999
-
[15]
McNutt, Paul Francoeur, Rishal Aggarwal, Tomohide Masuda, Rocco Meli, Matthew Ragoza, Jocelyn Sunseri, and David Ryan Koes
Andrew T. McNutt, Paul Francoeur, Rishal Aggarwal, Tomohide Masuda, Rocco Meli, Matthew Ragoza, Jocelyn Sunseri, and David Ryan Koes. GNINA 1.0: molecular docking with deep learning.Journal of Cheminformatics, 13:43, 2021
2021
-
[16]
Francoeur, Tomohide Masuda, Jocelyn Sunseri, Andrew Jia, Richard B
Paul G. Francoeur, Tomohide Masuda, Jocelyn Sunseri, Andrew Jia, Richard B. Iovanisci, Ian Snyder, and David Ryan Koes. Three-dimensional convolutional neural networks and a cross-docked data set for structure- based drug design.Journal of Chemical Information and Modeling, 60(9):4200–4215, 2020
2020
-
[17]
DiffDock: Diffusion steps, twists, and turns for molecular docking
Gabriele Corso, Hannes Stärk, Bowen Jing, Regina Barzilay, and Tommi Jaakkola. DiffDock: Diffusion steps, twists, and turns for molecular docking. InProceedings of the Eleventh International Conference on Learning Representations (ICLR), 2023
2023
-
[18]
Charifson, Joseph J
Paul S. Charifson, Joseph J. Corkery, Mark A. Murcko, and W. Patrick Walters. Consensus scoring: A method for obtaining improved hit rates from docking databases of three-dimensional structures into proteins.Journal of Medicinal Chemistry, 42(25):5100–5109, 1999
1999
-
[19]
Comparative evaluation of 11 scoring functions for molecular docking.Journal of Medicinal Chemistry, 46(12):2287–2303, 2003
Renxiao Wang, Yipin Lu, and Shaomeng Wang. Comparative evaluation of 11 scoring functions for molecular docking.Journal of Medicinal Chemistry, 46(12):2287–2303, 2003
2003
-
[20]
Kristal, and D
Jinn-Moon Yang, Yen-Fu Chen, Tsai-Wei Shen, Bruce S. Kristal, and D. Frank Hsu. Consensus scoring criteria for improving enrichment in virtual screening.Journal of Chemical Information and Modeling, 45(4):1134–1146, 2005. 66 AgenticPosesRanker: An Agentic AI Framework for Physically Grounded Ranking of Protein–Ligand Docking Poses
2005
-
[21]
ReAct: Synergizing reasoning and acting in language models
Shunyu Yao, Jeffrey Zhao, Dian Yu, Nan Du, Izhak Shafran, Karthik Narasimhan, and Yuan Cao. ReAct: Synergizing reasoning and acting in language models. InProceedings of the Eleventh International Conference on Learning Representations (ICLR), 2023
2023
-
[22]
Bran, Sam Cox, Oliver Schilter, Carlo Baldassari, Andrew D
Andres M. Bran, Sam Cox, Oliver Schilter, Carlo Baldassari, Andrew D. White, and Philippe Schwaller. Aug- menting large language models with chemistry tools.Nature Machine Intelligence, 6:525–535, 2024
2024
-
[23]
Boiko, Robert MacKnight, Ben Kline, and Gabe Gomes
Daniil A. Boiko, Robert MacKnight, Ben Kline, and Gabe Gomes. Autonomous chemical research with large language models.Nature, 624:570–578, 2023
2023
-
[24]
Chi, Quoc V
Jason Wei, Xuezhi Wang, Dale Schuurmans, Maarten Bosma, Brian Ichter, Fei Xia, Ed H. Chi, Quoc V . Le, and Denny Zhou. Chain-of-thought prompting elicits reasoning in large language models. InAdvances in Neural Information Processing Systems 35 (NeurIPS), 2022
2022
-
[25]
Towards understanding chain-of-thought prompting: An empirical study of what matters
Boshi Wang, Sewon Min, Xiang Deng, Jiaming Shen, You Wu, Luke Zettlemoyer, and Huan Sun. Towards understanding chain-of-thought prompting: An empirical study of what matters. InProceedings of the 61st Annual Meeting of the Association for Computational Linguistics (ACL), pages 2717–2739, 2023
2023
-
[26]
Faithful chain-of-thought reasoning
Qing Lyu, Shreya Havaldar, Adam Stein, Li Zhang, Delip Rao, Eric Wong, Marianna Apidianaki, and Chris Callison-Burch. Faithful chain-of-thought reasoning. InProceedings of the 13th International Joint Conference on Natural Language Processing and the 3rd Conference of the Asia-Pacific Chapter of the Association for Computational Linguistics (IJCNLP-AACL),...
2023
-
[27]
Miles Turpin, Julian Michael, Ethan Perez, and Samuel R. Bowman. Language models don’t always say what they think: Unfaithful explanations in chain-of-thought prompting. InAdvances in Neural Information Processing Systems, volume 36, 2023
2023
-
[28]
Measuring Faithfulness in Chain-of-Thought Reasoning
Tamera Lanham, Anna Chen, Ansh Radhakrishnan, Benoit Steiner, Carson Denison, Danny Hernandez, Dustin Li, Esin Durmus, Evan Hubinger, Deep Ganguli, et al. Measuring faithfulness in chain-of-thought reasoning.arXiv preprint arXiv:2307.13702, 2023
work page Pith review arXiv 2023
-
[29]
Liu, Kevin Lin, John Hewitt, Ashwin Paranjape, Michele Bevilacqua, Fabio Petroni, and Percy Liang
Nelson F. Liu, Kevin Lin, John Hewitt, Ashwin Paranjape, Michele Bevilacqua, Fabio Petroni, and Percy Liang. Lost in the middle: How language models use long contexts.Transactions of the Association for Computational Linguistics, 12:157–173, 2024
2024
-
[30]
Large language models are not robust multiple choice selectors
Chujie Zheng, Hao Zhou, Fandong Meng, Jie Zhou, and Minlie Huang. Large language models are not robust multiple choice selectors. InProceedings of the Twelfth International Conference on Learning Representations (ICLR), 2024
2024
-
[31]
Found in the middle: Permutation self-consistency improves listwise ranking in large language models
Raphael Tang, Crystina Zhang, Xueguang Ma, Jimmy Lin, and Ferhan Ture. Found in the middle: Permutation self-consistency improves listwise ranking in large language models. InProceedings of the 2024 Conference of the North American Chapter of the Association for Computational Linguistics (NAACL), pages 2327–2340, 2024
2024
-
[32]
The PDBbind database: collection of binding affinities for protein–ligand complexes with known three-dimensional structures.Journal of Medicinal Chemistry, 47(12):2977–2980, 2004
Renxiao Wang, Xueliang Fang, Yipin Lu, and Shaomeng Wang. The PDBbind database: collection of binding affinities for protein–ligand complexes with known three-dimensional structures.Journal of Medicinal Chemistry, 47(12):2977–2980, 2004
2004
-
[33]
The PDBbind database: methodologies and updates.Journal of Medicinal Chemistry, 48(12):4111–4119, 2005
Renxiao Wang, Xueliang Fang, Yipin Lu, Chao-Yie Yang, and Shaomeng Wang. The PDBbind database: methodologies and updates.Journal of Medicinal Chemistry, 48(12):4111–4119, 2005
2005
-
[34]
Brown, T
Lawrence D. Brown, T. Tony Cai, and Anirban DasGupta. Interval estimation for a binomial proportion.Statistical Science, 16(2):101–133, 2001
2001
-
[35]
Edwin B. Wilson. Probable inference, the law of succession, and statistical inference.Journal of the American Statistical Association, 22(158):209–212, 1927
1927
-
[36]
Version 2025.09.2
RDKit: Open-source cheminformatics.https://www.rdkit.org. Version 2025.09.2
2025
-
[37]
Streamlit: A faster way to build and share data apps, 2024
Streamlit, Inc. Streamlit: A faster way to build and share data apps, 2024. Accessed: 2025-01-15
2024
-
[38]
Burley, Jaroslav Koˇca, and Alexander S
David Sehnal, Sebastian Bittrich, Mandar Deshpande, Radka Svobodová, Karel Berka, Václav Bazgier, Sameer Velankar, Stephen K. Burley, Jaroslav Koˇca, and Alexander S. Rose. Mol* Viewer: modern web app for 3D visualization and analysis of large biomolecular structures.Nucleic Acids Research, 49(W1):W431–W437, 2021
2021
-
[39]
Joachim Haupt, Melissa F
Sebastian Salentin, Sven Schreiber, V . Joachim Haupt, Melissa F. Adasme, and Michael Schroeder. PLIP: fully automated protein–ligand interaction profiler.Nucleic Acids Research, 43(W1):W443–W447, 2015
2015
-
[40]
Adasme, Katja L
Melissa F. Adasme, Katja L. Linnemann, Sarah Naomi Bolz, Florian Kaiser, Sebastian Salentin, V . Joachim Haupt, and Michael Schroeder. PLIP 2021: expanding the scope of the protein–ligand interaction profiler to DNA and RNA.Nucleic Acids Research, 49(W1):W530–W534, 2021. 67 AgenticPosesRanker: An Agentic AI Framework for Physically Grounded Ranking of Pro...
2021
-
[41]
PLIP 2025: introducing protein- protein interactions to the protein-ligand interaction profiler.Nucleic Acids Research, 53(W1):W463–W465, 2025
Philipp Schake, Sarah Naomi Bolz, Katja Linnemann, and Michael Schroeder. PLIP 2025: introducing protein- protein interactions to the protein-ligand interaction profiler.Nucleic Acids Research, 53(W1):W463–W465, 2025
2025
-
[42]
Shrake and J
A. Shrake and J. A. Rupley. Environment and exposure to solvent of protein atoms. Lysozyme and insulin.Journal of Molecular Biology, 79(2):351–371, 1973
1973
-
[43]
Peter J. A. Cock, Tiago Antao, Jeffrey T. Chang, Brad A. Chapman, Cymon J. Cox, Andrew Dalke, Iddo Friedberg, Thomas Hamelryck, Frank Kauff, Bartek Wilczynski, and Michiel J. L. de Hoon. Biopython: freely available Python tools for computational molecular biology and bioinformatics.Bioinformatics, 25(11):1422–1423, 2009
2009
-
[44]
Thomas A. Halgren. Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94.Journal of Computational Chemistry, 17(5-6):490–519, 1996
1996
-
[45]
Bringing the MMFF force field to the RDKit: implementation and validation.Journal of Cheminformatics, 6:37, 2014
Paolo Tosco, Nikolaus Stiefl, and Gregory Landrum. Bringing the MMFF force field to the RDKit: implementation and validation.Journal of Cheminformatics, 6:37, 2014
2014
-
[46]
Charifson
Emanuele Perola and Paul S. Charifson. Conformational analysis of drug-like molecules bound to proteins: An extensive study of ligand reorganization upon binding.Journal of Medicinal Chemistry, 47(10):2499–2510, 2004
2004
-
[47]
Jeffrey.An Introduction to Hydrogen Bonding
George A. Jeffrey.An Introduction to Hydrogen Bonding. Oxford University Press, 1997
1997
-
[48]
The hydrogen bond in the solid state.Angewandte Chemie International Edition, 41(1):49–76, 2002
Thomas Steiner. The hydrogen bond in the solid state.Angewandte Chemie International Edition, 41(1):49–76, 2002
2002
-
[49]
Salt bridge stability in monomeric proteins.Journal of Molecular Biology, 293(5):1241–1255, 1999
Sandeep Kumar and Ruth Nussinov. Salt bridge stability in monomeric proteins.Journal of Molecular Biology, 293(5):1241–1255, 1999
1999
-
[50]
Hopkins, Colin R
Andrew L. Hopkins, Colin R. Groom, and Alexander Alex. Ligand efficiency: A useful metric for lead selection. Drug Discovery Today, 9(10):430–431, 2004
2004
-
[51]
Can LLMs express their uncertainty? An empirical evaluation of confidence elicitation in LLMs
Miao Xiong, Zhiyuan Hu, Xinyang Lu, Yifei Li, Jie Fu, Junxian He, and Bryan Hooi. Can LLMs express their uncertainty? An empirical evaluation of confidence elicitation in LLMs. InProceedings of the Twelfth International Conference on Learning Representations (ICLR), 2024
2024
-
[52]
Schrödinger, LLC. Maestro. Schrödinger, LLC, New York, NY . Molecular visualisation software. Used for structural figures of representative systems
-
[53]
Dahlgren, Ellery Russell, Christopher D
Chao Lu, Chuanjie Wu, Delaram Ghoreishi, Wei Chen, Lingle Wang, Wolfgang Damm, Markus K. Dahlgren, Ellery Russell, Christopher D. V on Bargen, Robert Abel, Richard A. Friesner, and Edward D. Harder. OPLS4: Improving force field accuracy on challenging regimes of chemical space.Journal of Chemical Theory and Computation, 17(7):4291–4300, 2021
2021
-
[54]
An ANI-2 enabled open-source protocol to estimate ligand strain after docking
Francois Berenger and Koji Tsuda. An ANI-2 enabled open-source protocol to estimate ligand strain after docking. Journal of Computational Chemistry, 46(1):e27478, 2024
2024
-
[55]
DSPy: Compiling Declarative Language Model Calls into Self-Improving Pipelines
Omar Khattab, Arnav Singhvi, Paridhi Maheshwari, Zhiyuan Zhang, Keshav Santhanam, Sri Vardhamanan, Saiful Haq, Ashutosh Sharma, et al. DSPy: Compiling declarative language model calls into self-improving pipelines. arXiv preprint arXiv:2310.03714, 2023
work page internal anchor Pith review arXiv 2023
-
[56]
Carlson, Richard D
Heather A. Carlson, Richard D. Smith, Kelly L. Damm-Ganamet, Jeanne A. Stuckey, Aqeel Ahmed, Maire A. Convery, Donald O. Somers, Michael Kranz, Patricia A. Elkins, Guanglei Cui, Catherine E. Peishoff, Millard H. Lambert, and James B. Dunbar, Jr. CSAR 2014: A benchmark exercise using unpublished data from pharma. Journal of Chemical Information and Modelin...
2014
-
[57]
Villarreal
Rodrigo Quiroga and Marcos A. Villarreal. Vinardo: A scoring function based on Autodock Vina improves scoring, docking, and virtual screening.PLoS ONE, 11(5):e0155183, 2016
2016
-
[58]
Ballester and John B
Pedro J. Ballester and John B. O. Mitchell. A machine learning approach to predicting protein–ligand binding affinity with applications to molecular docking.Bioinformatics, 26(9):1169–1175, 2010
2010
-
[59]
O’Boyle, Michael Banck, Craig A
Noel M. O’Boyle, Michael Banck, Craig A. James, Chris Morley, Tim Vandermeersch, and Geoffrey R. Hutchison. Open Babel: An open chemical toolbox.Journal of Cheminformatics, 3:33, 2011. 68
2011
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.