Recognition: unknown
Selecting optimal unrestricted Hartree-Fock trial wavefunctions for phaseless auxiliary-field quantum Monte Carlo: Accuracy and limitations in modeling three iron-sulfur clusters
Pith reviewed 2026-05-07 12:40 UTC · model grok-4.3
The pith
Chemical properties and physical symmetries, rather than variational energy, guide the choice of unrestricted Hartree-Fock trials for phaseless auxiliary-field quantum Monte Carlo on iron-sulfur clusters.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
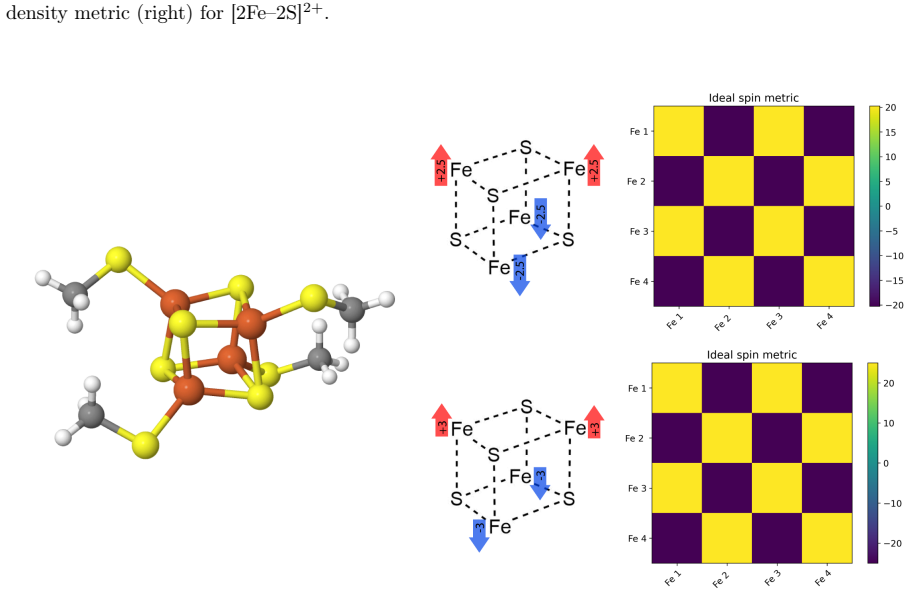
In active-space models of [2Fe-2S]²⁺, mixed-valent [4Fe-4S]²⁺, and [4Fe-4S]⁴⁺, the quality of ph-AFQMC ground-state energies depends on choosing UHF trials according to chemical properties and physical symmetries rather than variational energy; this selection yields accurate results even while the overlap with the trial vanishes, with the phaseless bias compensating for the induced sampling error toward negative energies.
What carries the argument
Unrestricted Hartree-Fock trial wavefunction chosen according to chemical properties and physical symmetries, used as the importance function in phaseless auxiliary-field quantum Monte Carlo.
If this is right
- Accurate ground-state energies become accessible for these strongly correlated iron-sulfur clusters using inexpensive mean-field trials.
- Mean-field wavefunctions remain viable importance functions for ph-AFQMC when selected by symmetry rather than energy.
- The phaseless approximation can offset sampling biases in cases where the trial overlap vanishes.
- The same symmetry-based selection principle applies when choosing reference states for coupled-cluster calculations on similar systems.
Where Pith is reading between the lines
- For other transition-metal clusters, trial selection by matching physical symmetries may systematically improve ph-AFQMC results over energy minimization.
- The rapid loss of overlap signals a need for improved importance functions or alternative constraints that reduce dependence on phaseless bias compensation.
- Similar sampling-bias issues could appear in other quantum Monte Carlo or coupled-cluster applications that rely on mean-field references for strongly correlated states.
Load-bearing premise
The phaseless constraint reliably compensates for sampling bias from vanishing overlap with the UHF trial without introducing uncontrolled errors in these specific active-space models.
What would settle it
A direct comparison showing that ph-AFQMC energies with symmetry-guided UHF trials deviate substantially from exact benchmarks or high-accuracy reference values for any of the three clusters would falsify the accuracy claim.
Figures











read the original abstract
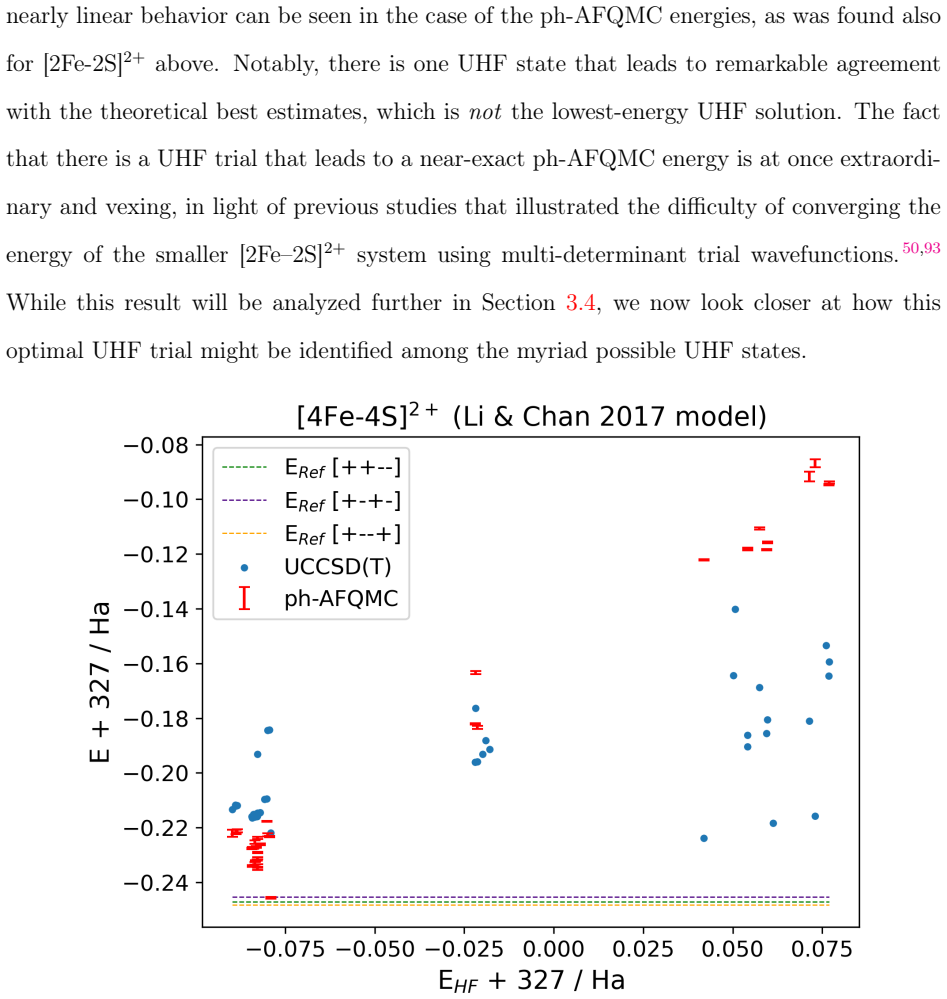
Phaseless auxiliary-field quantum Monte Carlo (ph-AFQMC) has emerged as a promising electronic structure method for correlated electronic systems. However, the quality of its predictions depends critically on the choice of trial wavefunction, and it is not obvious how to make an optimal choice especially for strongly correlated states of large systems. Mean-field wavefunctions are compelling trial wavefunction candidates as they map directly to chemical concepts and can be obtained with $O(N^4)$ cost. Yet in the strongly correlated regime one faces a symmetry dilemma and the existence of multiple nearly-degenerate solutions. In this work we investigate active space models of [2Fe-2S]$^{2+}$, mixed-valent [4Fe-4S]$^{2+}$, and [4Fe-4S]$^{4+}$ and explore the sensitivity of ph-AFQMC to the choice of unrestricted Hartree-Fock trial wavefunction. We find that chemical properties and physical symmetries, rather than the variational energy, ought to guide the choice of mean-field trial for ph-AFQMC (or reference state for coupled cluster models), and show that surprisingly accurate ground-state energies for these systems can be obtained. However, in all cases we find a rapidly vanishing overlap between the stochastic wavefunction and the UHF trial, indicating that the trials are suboptimal importance functions. By analogy to a similar situation in the stretched helium dimer cation, we show how this sampling bias pushes ph-AFQMC towards artificially negative energies, which evidently can be compensated for by the phaseless bias in certain cases.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript examines the sensitivity of phaseless auxiliary-field quantum Monte Carlo (ph-AFQMC) to the choice of unrestricted Hartree-Fock (UHF) trial wavefunctions for active-space models of three iron-sulfur clusters ([2Fe-2S]^{2+}, mixed-valent [4Fe-4S]^{2+}, and [4Fe-4S]^{4+}). It concludes that chemical properties and physical symmetries, rather than the variational energy of the trial, should guide selection of the mean-field trial (or reference state for coupled cluster), yielding surprisingly accurate ground-state energies. The authors report that overlap with all UHF trials vanishes rapidly, inducing a sampling bias that drives energies artificially negative (illustrated via stretched He2+ analogy), but state that this bias is evidently compensated by the phaseless constraint in the cases studied.
Significance. If the accuracy claims and bias-compensation observation hold, the work offers practical guidance for trial selection in ph-AFQMC for strongly correlated transition-metal systems and highlights the limitations of UHF importance functions due to vanishing overlap. The emphasis on symmetry over energy minimization could influence reference-state choices in related methods, though the post-hoc nature of the compensation argument limits its immediate generality.
major comments (3)
- [Abstract] Abstract: the claim that the phaseless bias 'evidently can be compensated' for the sampling bias induced by vanishing UHF overlap rests on a post-hoc interpretation and the stretched-He2+ analogy without a direct quantitative test or residual-error bound specific to the iron-sulfur active-space models.
- [Results for the iron-sulfur clusters] Results sections on the three clusters: the size of the active spaces, orbital selection, and convergence of the ph-AFQMC energies (walker number, time step, etc.) are not detailed, so it is not possible to assess whether the reported agreement with reference data is robust or affected by uncontrolled approximations.
- [Discussion of trial selection] Discussion of trial selection: because overlap vanishes rapidly for every UHF trial examined, the recommendation to prioritize symmetries over variational energy lacks a controlled comparison demonstrating that energy-minimized trials systematically underperform; the observed accuracy may reflect case-specific cancellation rather than a general principle.
minor comments (1)
- [Results] The manuscript would benefit from a summary table listing the UHF solutions, their symmetries, variational energies, and resulting ph-AFQMC energies for each cluster to facilitate direct comparison.
Simulated Author's Rebuttal
We thank the referee for the detailed and constructive report. The comments highlight important points regarding the strength of our claims, the need for methodological details, and the generality of our trial-selection guidance. We address each major comment below and have revised the manuscript accordingly to improve clarity and rigor.
read point-by-point responses
-
Referee: [Abstract] Abstract: the claim that the phaseless bias 'evidently can be compensated' for the sampling bias induced by vanishing UHF overlap rests on a post-hoc interpretation and the stretched-He2+ analogy without a direct quantitative test or residual-error bound specific to the iron-sulfur active-space models.
Authors: We agree that the original phrasing in the abstract was too strong and could be read as post-hoc. The stretched He2+ example is intended only as a mechanistic illustration of how vanishing overlap induces a sampling bias toward lower energies; the iron-sulfur results themselves show that the final ph-AFQMC energies remain close to reference values despite this bias. We have revised the abstract to replace 'evidently can be compensated' with 'appears to be offset by the phaseless constraint in the systems examined,' added a brief caveat that a general quantitative bound is not provided, and inserted a forward-looking sentence noting that systematic error analysis remains an open question for future work. revision: yes
-
Referee: [Results for the iron-sulfur clusters] Results sections on the three clusters: the size of the active spaces, orbital selection, and convergence of the ph-AFQMC energies (walker number, time step, etc.) are not detailed, so it is not possible to assess whether the reported agreement with reference data is robust or affected by uncontrolled approximations.
Authors: This observation is correct; the original manuscript emphasized trial-wavefunction sensitivity at the expense of explicit technical specifications. We have added a dedicated subsection (now Section 2.2) that reports the active-space dimensions (CAS(10,10) for [2Fe-2S]^{2+}, CAS(20,20) for the [4Fe-4S] clusters), the orbital-selection protocol based on natural-orbital occupation thresholds, and convergence data with respect to walker population (up to 10^4 walkers), imaginary-time step (0.005–0.01 a.u.), and projection length. Statistical error bars and autocorrelation times are now tabulated, confirming that the quoted energies are converged within the reported uncertainties. revision: yes
-
Referee: [Discussion of trial selection] Discussion of trial selection: because overlap vanishes rapidly for every UHF trial examined, the recommendation to prioritize symmetries over variational energy lacks a controlled comparison demonstrating that energy-minimized trials systematically underperform; the observed accuracy may reflect case-specific cancellation rather than a general principle.
Authors: We partially concur. While overlap decay is universal across the UHF solutions we tested, we did compare trials that preserve versus break the expected spin and point-group symmetries. In each cluster the symmetry-preserving (higher-variational-energy) trials produced ph-AFQMC energies closer to the reference values than the lowest-energy symmetry-broken solutions. Nevertheless, we acknowledge that this is not an exhaustive scan over all possible mean-field solutions and that some degree of error cancellation specific to these active-space models cannot be ruled out. The revised discussion now frames the recommendation as an empirical guideline supported by the trends in the three clusters studied, explicitly cautions against assuming universality, and suggests that future benchmarks on additional systems would be valuable. revision: partial
Circularity Check
No circularity: empirical computational survey with external analogy
full rationale
The manuscript is a computational survey that compares multiple UHF trial wavefunctions for ph-AFQMC on three active-space iron-sulfur models, reporting energies, overlaps, and symmetry properties against reference data. No derivation chain exists that reduces a claimed result to its own inputs by construction; the central recommendation follows from direct numerical comparisons rather than from any fitted parameter renamed as a prediction or from a self-citation that supplies the uniqueness or ansatz. The noted vanishing overlap and bias compensation are presented as observations (with an external stretched-He2+ analogy), not as a self-referential proof. The paper therefore remains self-contained against external benchmarks.
Axiom & Free-Parameter Ledger
axioms (2)
- domain assumption Unrestricted Hartree-Fock provides a usable mean-field trial wavefunction whose symmetry properties can be meaningfully compared to chemical intuition.
- domain assumption The phaseless constraint in AFQMC can partially cancel sampling bias without introducing uncontrolled systematic error for the chosen active spaces.
Reference graph
Works this paper leans on
-
[1]
The Journal of Physical Chemistry A , author =
Study of. The Journal of Physical Chemistry A , author =. 2022 , pages =. doi:10.1021/acs.jpca.1c10354 , language =
-
[2]
Can phaseless auxiliary-field quantum
-
[3]
The. Chemical Reviews , author =. 2024 , pages =. doi:10.1021/acs.chemrev.3c00755 , language =
-
[4]
Accounts of Chemical Research , author =
Thermally. Accounts of Chemical Research , author =. 2018 , pages =. doi:10.1021/acs.accounts.8b00174 , language =
-
[5]
The Journal of Chemical Physics , author =
The performance of phaseless auxiliary-field quantum. The Journal of Chemical Physics , author =. 2020 , pages =. doi:10.1063/5.0024835 , language =
-
[6]
Journal of Chemical Theory and Computation , author =
Either. Journal of Chemical Theory and Computation , author =. 2022 , pages =. doi:10.1021/acs.jctc.2c00905 , language =
-
[7]
Comparative study of. Heliyon , author =. 2024 , pages =. doi:10.1016/j.heliyon.2024.e30926 , language =
-
[8]
The Journal of Physical Chemistry A , author =
Benchmarking. The Journal of Physical Chemistry A , author =. 2024 , pages =. doi:10.1021/acs.jpca.4c03273 , language =
-
[9]
The Journal of Physical Chemistry A , author =
Origin of the. The Journal of Physical Chemistry A , author =. 2022 , pages =. doi:10.1021/acs.jpca.1c10492 , language =
-
[10]
The Journal of Chemical Physics , author =
Equation-of-motion spin-flip coupled-cluster model with single and double substitutions:. The Journal of Chemical Physics , author =. 2004 , pages =. doi:10.1063/1.1630018 , abstract =
-
[11]
The Journal of Chemical Physics , author =
Benchmarking the performance of time-dependent density functional methods , volume =. The Journal of Chemical Physics , author =. 2012 , pages =. doi:10.1063/1.3689445 , abstract =
-
[12]
Journal of Chemical Theory and Computation , author =
Optimization of the. Journal of Chemical Theory and Computation , author =. 2025 , pages =. doi:10.1021/acs.jctc.5c00345 , language =
-
[13]
Unitary. Physica Scripta , author =. 1980 , pages =. doi:10.1088/0031-8949/21/3-4/012 , language =
-
[14]
The Journal of Physical Chemistry Letters , author =
The. The Journal of Physical Chemistry Letters , author =. 2020 , pages =. doi:10.1021/acs.jpclett.0c02621 , abstract =
-
[15]
The Journal of Chemical Physics , author =
The performance of. The Journal of Chemical Physics , author =. 2020 , pages =. doi:10.1063/5.0027617 , abstract =
-
[16]
Journal of Chemical Theory and Computation , author =
The. Journal of Chemical Theory and Computation , author =. 2022 , pages =. doi:10.1021/acs.jctc.1c00830 , abstract =
-
[17]
Patel, Smik and Jayakumar, Praveen and Huang, Rick and Zeng, Tao and Izmaylov, Artur F. , month = dec, year =. Quantum. doi:10.48550/arXiv.2509.01061 , abstract =
-
[18]
Journal of Chemical Theory and Computation , author =
Multistate. Journal of Chemical Theory and Computation , author =. 2026 , pages =. doi:10.1021/acs.jctc.5c01849 , abstract =
-
[19]
Multireference. Chemical Reviews , author =. 2026 , pages =. doi:10.1021/acs.chemrev.5c00866 , abstract =
-
[20]
Nature Communications , author =
Numerically exact configuration interaction at quadrillion-determinant scale , volume =. Nature Communications , author =. 2025 , pages =. doi:10.1038/s41467-025-65967-7 , language =
-
[21]
Chemical Physics Reviews , author =
Small tensor product distributed active space (. Chemical Physics Reviews , author =. 2024 , pages =. doi:10.1063/5.0227122 , abstract =
-
[22]
Variational quantum computation of excited states
Variational. Quantum , author =. 2019 , pages =. doi:10.22331/q-2019-07-01-156 , abstract =
-
[23]
Physical Review Research , author =
Quantum equation of motion for computing molecular excitation energies on a noisy quantum processor , volume =. Physical Review Research , author =. 2020 , pages =. doi:10.1103/PhysRevResearch.2.043140 , language =
-
[24]
The Journal of Physical Chemistry A , author =
Unitary. The Journal of Physical Chemistry A , author =. 2023 , pages =. doi:10.1021/acs.jpca.3c02781 , language =
-
[25]
Journal of Chemical Theory and Computation , author =
Generalized. Journal of Chemical Theory and Computation , author =. 2019 , pages =. doi:10.1021/acs.jctc.8b01004 , language =
-
[26]
Quantum Science and Technology , author =
Strategies for quantum computing molecular energies using the unitary coupled cluster ansatz , volume =. Quantum Science and Technology , author =. 2019 , pages =. doi:10.1088/2058-9565/aad3e4 , abstract =
-
[27]
The theory of variational hybrid quantum-classical algorithms,
The theory of variational hybrid quantum-classical algorithms , volume =. New Journal of Physics , author =. 2016 , pages =. doi:10.1088/1367-2630/18/2/023023 , number =
-
[28]
O'Malley, P. J. J. and Babbush, R. and Kivlichan, I. D. and Romero, J. and McClean, J. R. and Barends, R. and Kelly, J. and Roushan, P. and Tranter, A. and Ding, N. and Campbell, B. and Chen, Y. and Chen, Z. and Chiaro, B. and Dunsworth, A. and Fowler, A. G. and Jeffrey, E. and Lucero, E. and Megrant, A. and Mutus, J. Y. and Neeley, M. and Neill, C. and Q...
-
[29]
npj Quantum Information , author =
Exact electronic states with shallow quantum circuits from global optimisation , volume =. npj Quantum Information , author =. 2023 , pages =. doi:10.1038/s41534-023-00744-2 , abstract =
-
[30]
An adaptive variational algorithm for exact molecular simulations on a quantum computer , volume =. Nature Communications , author =. 2019 , pages =. doi:10.1038/s41467-019-10988-2 , abstract =
-
[31]
Photochemical. Molecular Biology , author =. 2024 , pages =. doi:10.1134/S0026893324010047 , abstract =
-
[32]
Towards quantum chemistry on a quantum computer , volume =. Nature Chemistry , author =. 2010 , pages =. doi:10.1038/nchem.483 , language =
-
[33]
Chemistry – An Asian Journal , author =
Evolution of. Chemistry – An Asian Journal , author =. 2025 , pages =. doi:10.1002/asia.202401291 , abstract =
-
[34]
Comprehensive. Chemical Reviews , author =. 2025 , pages =. doi:10.1021/acs.chemrev.5c00021 , language =
-
[35]
Journal of Chemical Theory and Computation , author =
Quantum. Journal of Chemical Theory and Computation , author =. 2024 , pages =. doi:10.1021/acs.jctc.4c01071 , language =
-
[36]
Variational benchmarks for quantum many-body problems , volume =. Science , author =. 2024 , pages =. doi:10.1126/science.adg9774 , abstract =
-
[37]
Qubit coupled cluster method: A systematic approach to quantum chemistry on a quantum computer,
Qubit. Journal of Chemical Theory and Computation , author =. 2018 , pages =. doi:10.1021/acs.jctc.8b00932 , abstract =
-
[38]
Jenab, Seyyed Mehdi Hosseini and Henderson, Brandon and Genin, Scott N. , month = mar, year =. Parallel. doi:10.48550/arXiv.2603.08883 , abstract =
-
[39]
Journal of Chemical Theory and Computation , author =
Correlated-. Journal of Chemical Theory and Computation , author =. 2016 , pages =. doi:10.1021/acs.jctc.6b00569 , abstract =
-
[40]
Smeyers, Yves G. , year =. The half projected hartree-fock model for determining singlet excited states , volume =. Advances in. doi:10.1016/s0065-3276(08)60486-4 , abstract =
-
[41]
Galván, Ignacio and Lindh, Roland and Li Manni, Giovanni , month = mar, year =
Song, Maru and Bonfirraro, Luca and Fdez. Galván, Ignacio and Lindh, Roland and Li Manni, Giovanni , month = mar, year =. Spin-. doi:10.26434/chemrxiv.15000528/v2 , abstract =
-
[42]
Journal of the American Chemical Society , author =
Activation of. Journal of the American Chemical Society , author =. 2024 , pages =. doi:10.1021/jacs.4c13490 , language =
-
[43]
Journal of the American Chemical Society , author =
Periodic. Journal of the American Chemical Society , author =. 2025 , pages =. doi:10.1021/jacs.5c09551 , language =
-
[44]
The Journal of Chemical Physics , author =
The electronic complexity of the ground-state of the. The Journal of Chemical Physics , author =. 2019 , pages =. doi:10.1063/1.5063376 , abstract =
-
[45]
Svore, Dave Wecker, and Matthias Troyer
Elucidating reaction mechanisms on quantum computers , volume =. Proceedings of the National Academy of Sciences , author =. 2017 , pages =. doi:10.1073/pnas.1619152114 , abstract =
-
[46]
Journal of Chemical Theory and Computation , author =
A. Journal of Chemical Theory and Computation , author =. 2025 , pages =. doi:10.1021/acs.jctc.5c01038 , language =
-
[47]
Inorganic Chemistry , author =
Vibrational. Inorganic Chemistry , author =. 2025 , pages =. doi:10.1021/acs.inorgchem.5c02572 , language =
-
[48]
Proceedings of the National Academy of Sciences , author =
Synthetic analogues of [. Proceedings of the National Academy of Sciences , author =. 2011 , pages =. doi:10.1073/pnas.1106472108 , abstract =
-
[49]
The Journal of Physical Chemistry A , author =
A-. The Journal of Physical Chemistry A , author =. 2023 , pages =. doi:10.1021/acs.jpca.3c04977 , language =
-
[50]
Chemistry beyond the scale of exact diagonalization on a quantum-centric supercomputer , volume =. Science Advances , author =. 2025 , pages =. doi:10.1126/sciadv.adu9991 , abstract =
-
[51]
Communications Chemistry , author =
Convex. Communications Chemistry , author =. 2026 , pages =. doi:10.1038/s42004-025-01842-2 , abstract =
-
[52]
Tuning (. ChemPhysChem , author =. 2024 , pages =. doi:10.1002/cphc.202300791 , abstract =
-
[53]
and Walton, Richard A
Cotton, Frank Albert and Murillo, Carlos A. and Walton, Richard A. , year =. Multiple bonds between metal atoms , isbn =
-
[54]
doi:10.1007/b99204 , abstract =
Personality in. doi:10.1007/b99204 , abstract =
-
[55]
Sakurai, J. J. and Napolitano, Jim , month = sep, year =. Modern. doi:10.1017/9781108587280 , abstract =
-
[56]
Journal of Chemical Theory and Computation , author =
Critical. Journal of Chemical Theory and Computation , author =. 2025 , pages =. doi:10.1021/acs.jctc.5c00375 , abstract =
-
[57]
Programmable simulations of molecules and materials with reconfigurable quantum processors , volume =. Nature Physics , author =. 2025 , pages =. doi:10.1038/s41567-024-02738-z , language =
-
[58]
The Journal of Physical Chemistry Letters , author =
Challenges and. The Journal of Physical Chemistry Letters , author =. 2025 , pages =. doi:10.1021/acs.jpclett.5c01680 , abstract =
-
[59]
Electronic Structure , author =
Subspace methods for electronic structure simulations on quantum computers , volume =. Electronic Structure , author =. 2024 , pages =. doi:10.1088/2516-1075/ad3592 , abstract =
-
[60]
Classical solution of the FeMo-cofactor model to chemical accuracy and its implications
Zhai, Huanchen and Li, Chenghan and Zhang, Xing and Li, Zhendong and Lee, Seunghoon and Chan, Garnet Kin-Lic , month = jan, year =. Classical solution of the. doi:10.48550/arXiv.2601.04621 , abstract =
-
[61]
Ku, Calvin and Chen, Yu-Cheng and Hu, Alice and Hsieh, Min-Hsiu , month = oct, year =. Benchmarking. doi:10.48550/arXiv.2510.01710 , abstract =
-
[62]
Polynomial-time quantum algorithm for the simulation of chemical dynamics,
Polynomial-time quantum algorithm for the simulation of chemical dynamics , volume =. Proceedings of the National Academy of Sciences , author =. 2008 , pages =. doi:10.1073/pnas.0808245105 , abstract =
-
[63]
Quantum algorithms for fermionic simulations , volume =. Physical Review A , author =. 2001 , pages =. doi:10.1103/PhysRevA.64.022319 , language =
-
[64]
Simulating. Physical Review Letters , author =. 2015 , pages =. doi:10.1103/PhysRevLett.114.090502 , language =
-
[65]
Sze, Michelle Wynne and Manrique, David Zsolt and Ramo, David Muñoz and Fitzpatrick, Nathan , month = nov, year =. Shorter width truncated. doi:10.48550/arXiv.2511.09461 , abstract =
-
[66]
Asthana, Ayush , month = dec, year =. Quantum. doi:10.48550/arXiv.2512.11788 , abstract =
-
[67]
Quantum. PRX Quantum , author =. 2021 , note =. doi:10.1103/PRXQuantum.2.040352 , abstract =
-
[68]
Parrish, Robert M. and McMahon, Peter L. , month = sep, year =. Quantum. doi:10.48550/arXiv.1909.08925 , abstract =
-
[69]
Identification of a spin-coupled. Chem. Sci. , author =. 2014 , pages =. doi:10.1039/C4SC00337C , abstract =
-
[70]
Journal of the American Chemical Society , author =. 2001 , pages =. doi:10.1021/ja011860y , abstract =
-
[71]
International Journal of Quantum Chemistry , author =
Influence of the protein and. International Journal of Quantum Chemistry , author =. 2018 , pages =. doi:10.1002/qua.25627 , abstract =
-
[72]
Simulating physics with computers,
Simulating physics with computers , volume =. International Journal of Theoretical Physics , author =. 1982 , pages =. doi:10.1007/BF02650179 , language =
-
[73]
, translator =
Kitaev, Aleksej Ju and Šen, Aleksandr Ch and Vjalyj, Michail N. , translator =. Classical and quantum computation , isbn =
-
[74]
Simulated. Science , author =. 2005 , pages =. doi:10.1126/science.1113479 , abstract =
-
[75]
Journal of Computational Physics , author =
Solution of the. Journal of Computational Physics , author =. 1982 , pages =. doi:10.1016/0021-9991(82)90091-2 , language =
-
[76]
and Chuang, Isaac L
Nielsen, Michael A. and Chuang, Isaac L. , year =. Quantum computation and quantum information , isbn =
-
[77]
Kottmann, and Alán Aspuru-Guzik.Quantum Computing for Quantum Chemistry
Schleich, Philipp and Calderón, Luis Mantilla and Sun, Chong and Bagherimehrab, Mohsen and Aldossary, Abdulrahman and Kottmann, Jakob S. and Aspuru-Guzik, Alán , month = jun, year =. Quantum. doi:10.1021/acsinfocus.7e9012 , language =
-
[78]
Simulation of electronic structure. Molecular Physics , author =. 2011 , pages =. doi:10.1080/00268976.2011.552441 , language =
-
[79]
Genin, Scott N. and Kwon, Ohyun and Jenab, Seyyed Mehdi Hosseini and Lim, Seon-Jeong and Kim, Taehyung and Kim, Tae-Gon and Gherib, Rami and Harper, Angela F. and Ryabinkin, Ilya G. and Helander, Michael G. , month = dec, year =. Towards. doi:10.48550/arXiv.2512.13657 , abstract =
-
[80]
Effect of size, charge, and spin state on. Chemical Science , author =. 2025 , pages =. doi:10.1039/D4SC08225G , abstract =
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.