Recognition: unknown
CDFCI: High-Performance Parallel Software for Many-Body Large-Scale Eigenvalue Problems
Pith reviewed 2026-05-08 15:40 UTC · model grok-4.3
The pith
CDFCI computes low-lying eigenpairs of large fermionic Hamiltonians with state-of-the-art accuracy and competitive parallel speed.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
CDFCI implements a coordinate-descent-based selected configuration interaction method together with dedicated parallelization, attaining state-of-the-art accuracy and competitive performance on representative quantum chemistry and condensed matter test cases relative to established SCI and DMRG codes.
What carries the argument
Coordinate-descent selected configuration interaction algorithm combined with shared-memory parallelization strategies that distribute the configuration search and matrix operations across cores.
If this is right
- Users can solve larger active-space problems in ab initio electronic structure without switching to specialized hardware.
- The same code handles both molecular and lattice Hamiltonians within one framework.
- Integration with PySCF allows the method to slot directly into existing many-body workflows.
- Open-source release and documentation lower the barrier for community testing on new model systems.
Where Pith is reading between the lines
- The coordinate-descent approach may extend to time-dependent or driven problems if the iteration is adapted to include external fields.
- Performance on current multi-core chips suggests the method could scale further with GPU offloading or distributed-memory versions.
- Because the algorithm avoids full diagonalization, it might pair with embedding techniques to treat even bigger systems at reduced cost.
Load-bearing premise
The parallel coordinate-descent procedure maintains full accuracy while avoiding hidden overheads that would erase its speed advantage on the broad class of non-relativistic fermionic Hamiltonians.
What would settle it
A benchmark on a representative Hamiltonian where CDFCI either deviates from the reference energies obtained by CIPSI, SHCI, or DMRG by more than the stated tolerance or runs slower than those codes on identical multi-core hardware.
Figures
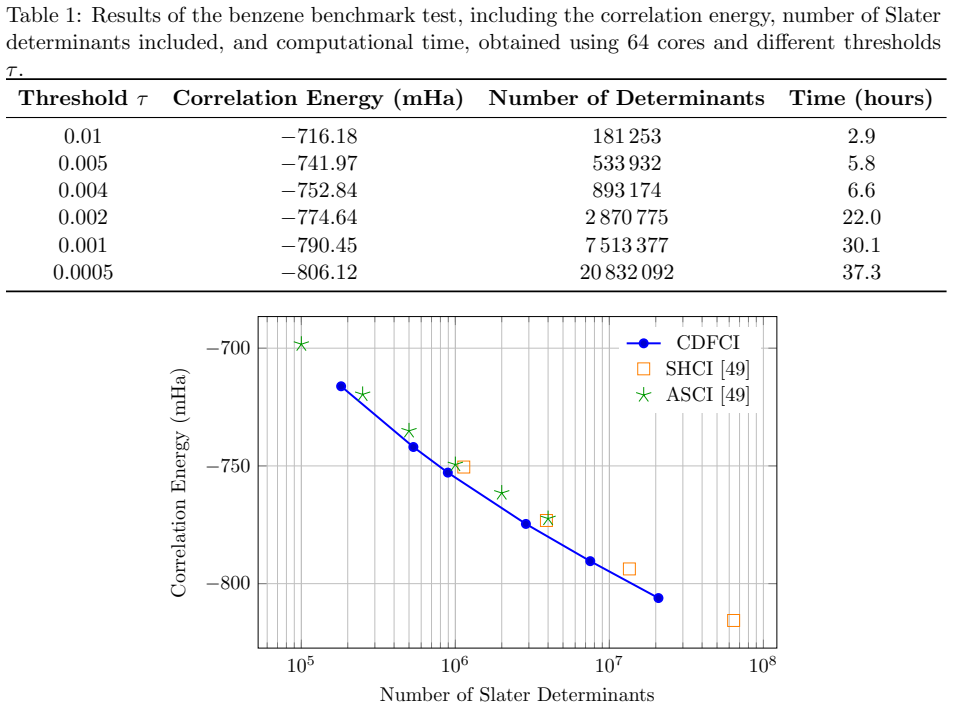
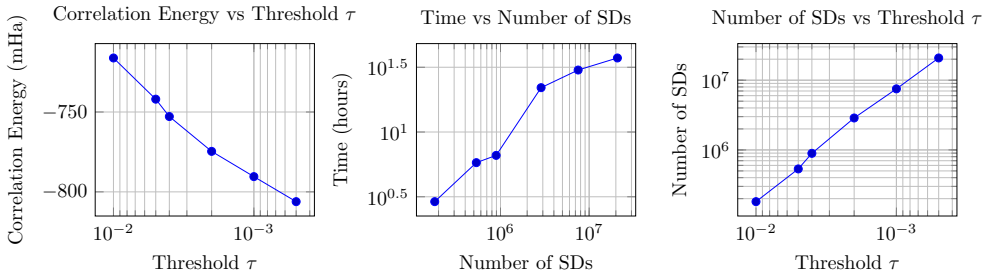

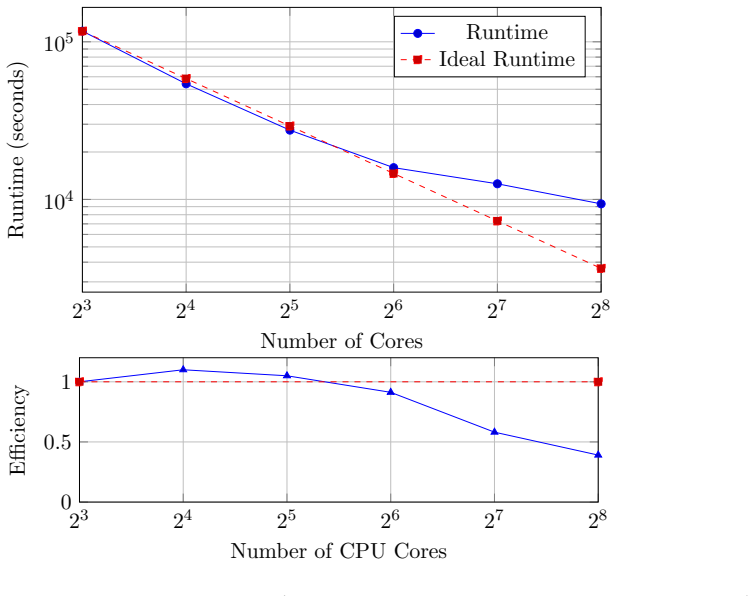
read the original abstract
CDFCI is a shared-memory parallel numerical program for computing low-lying eigenpairs of large-scale, non-relativistic fermionic Hamiltonians. The software is designed to handle a broad class of many-body quantum models, including both ab initio electronic structure Hamiltonians and lattice-based Hamiltonians arising in condensed matter physics. CDFCI combines an efficient coordinate-descent-based selected configuration interaction algorithm with dedicated parallelization strategies, achieving high performance on modern multi-core architectures. Benchmark results on representative quantum chemistry and condensed matter test cases demonstrate that CDFCI attains state-of-the-art accuracy with competitive performance compared to established selected configuration interaction (such as CIPSI or SHCI) and DMRG implementations. The software is open-source, extensively documented, and provides a Python interface for seamless integration with PySCF and other many-body simulation workflows.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript presents CDFCI, a shared-memory parallel software package implementing a coordinate-descent-based selected configuration interaction (SCI) algorithm for computing low-lying eigenpairs of large-scale non-relativistic fermionic Hamiltonians. It targets both ab initio quantum chemistry and lattice models from condensed matter physics, with dedicated parallelization strategies for modern multi-core hardware. Benchmark results on representative test cases are reported to demonstrate state-of-the-art accuracy and competitive performance relative to established methods such as CIPSI, SHCI, and DMRG. The software is open-source, documented, and provides a Python interface for integration with PySCF and similar workflows.
Significance. If the benchmark results and implementation details hold, CDFCI would be a significant contribution to computational many-body physics by offering an efficient, scalable tool for challenging eigenvalue problems that arise in electronic structure and lattice Hamiltonians. The combination of the coordinate-descent SCI approach with explicit parallel strategies addresses practical performance needs on contemporary hardware, and the open-source release with PySCF integration promotes reproducibility and broader adoption. The absence of hidden overheads or accuracy trade-offs in the reported tests supports the design goals without apparent internal inconsistencies.
minor comments (2)
- The abstract would benefit from inclusion of at least one quantitative benchmark metric (e.g., a specific energy error or wall-time comparison) to better substantiate the 'state-of-the-art accuracy' and 'competitive performance' claims for readers who encounter only the abstract.
- In the implementation or parallelization section, a brief pseudocode snippet or diagram illustrating the load-balancing and communication pattern would improve clarity for users adapting the code to new Hamiltonians.
Simulated Author's Rebuttal
We thank the referee for their positive and constructive review of our manuscript. We are pleased that the referee recognizes CDFCI as a significant contribution to computational many-body physics and recommends acceptance. The report contains no major comments requiring specific responses or revisions.
Circularity Check
No circularity detected in derivation or claims
full rationale
The paper describes a software implementation of a coordinate-descent selected CI algorithm with parallelization for fermionic eigenvalue problems. Its central claims consist of benchmarked accuracy and performance on external test cases compared to CIPSI, SHCI, and DMRG. These are empirical validations against independent implementations rather than internal predictions, fitted parameters renamed as outputs, or self-referential definitions. No load-bearing step reduces by construction to the paper's own inputs or prior self-citations; the algorithm description and parallel strategies are presented as design choices whose efficacy is measured externally. The derivation chain is therefore self-contained and non-circular.
Axiom & Free-Parameter Ledger
axioms (1)
- domain assumption Coordinate-descent selected configuration interaction remains accurate and efficient for the broad class of non-relativistic fermionic Hamiltonians when combined with the described parallelization.
Reference graph
Works this paper leans on
-
[1]
Davidson
Ernest R. Davidson. The iterative calculation of a few of the lowest eigenvalues and corre- sponding eigenvectors of large real-symmetric matrices.Journal of Computational Physics, 17:87–94, 1975
1975
-
[2]
Møller and M
Chr. Møller and M. S. Plesset. Note on an approximation treatment for many-electron systems. Physical Review, 46:618–622, 1934
1934
-
[3]
J. A. Pople, R. Seeger, and R. Krishnan. Variational Configuration Interaction methods and comparison with perturbation theory.International Journal of Quantum Chemistry, S4:149– 163, 1977
1977
-
[4]
Huron, J.-P
B. Huron, J.-P. Malrieu, and P. Rancurel. Iterative perturbation calculations of ground and excited state energies from multiconfigurational zeroth-order wavefunctions.The Journal of Chemical Physics, 58:5745–5759, 1973
1973
-
[5]
Tubman, C
Norm M. Tubman, C. Daniel Freeman, Daniel S. Levine, Diptarka Hait, Martin Head-Gordon, and K. Birgitta Whaley. Modern approaches to exact diagonalization and selected Configu- ration Interaction with the adaptive sampling CI method.Journal of Chemical Theory and Computation, 16(4):2139–2159, April 2020
2020
-
[6]
Kyeong Su Min and Jae Woo Park. Second-order Complete Active Space Perturbation Theory (CASPT2) andN-Electron Valence state Perturbation Theory (NEVPT2) based on Adaptive Sampling Configuration Interaction Self-Consistent Field (ASCI-SCF).Journal of Chemical Theory and Computation, 21(11):5425–5436, June 2025
2025
-
[7]
Holmes, Nathan M
Adam A. Holmes, Nathan M. Tubman, and C. J. Umrigar. Heat-bath Configuration Interac- tion: An efficient selected Configuration Interaction algorithm inspired by heat-bath sampling. Journal of Chemical Theory and Computation, 12(8):3674–3680, 2016
2016
-
[8]
Mihkel Ugandi and Michael Roemelt. A configuration-based heatbath-CI for spin-adapted multireference electronic structure calculations with large active spaces.Journal of Computational Chemistry, 44(31):2374–2390, 2023. eprint: https://onlinelibrary.wiley.com/doi/pdf/10.1002/jcc.27203
-
[9]
Holmes, Gregory Jeanmairet, Ali Alavi, and C
Sandeep Sharma, Adam A. Holmes, Gregory Jeanmairet, Ali Alavi, and C. J. Umrigar. Semis- tochastic heat-bath Configuration Interaction method: Selected Configuration Interaction with semistochastic perturbation theory.Journal of Chemical Theory and Computation, 13(4):1595– 1604, 2017. 28
2017
-
[10]
Relativistic Semistochastic Heat-Bath Configuration Inter- action.Journal of Chemical Theory and Computation, 19(3):848–855, February 2023
Xubo Wang and Sandeep Sharma. Relativistic Semistochastic Heat-Bath Configuration Inter- action.Journal of Chemical Theory and Computation, 19(3):848–855, February 2023
2023
-
[11]
Hoffmann
Ning Zhang, Wenjian Liu, and Mark R. Hoffmann. Iterative Configuration Interaction with selection.Journal of Chemical Theory and Computation, 16(4):2296–2316, April 2020. Pub- lisher: American Chemical Society
2020
-
[12]
Greene, Robert J
Samuel M. Greene, Robert J. Webber, Jonathan Weare, and Timothy C. Berkelbach. Improved fast randomized iteration approach to full Configuration Interaction.Journal of Chemical Theory and Computation, 16(9):5572–5585, sep 2020
2020
-
[13]
Goings, Hang Hu, Chao Yang, and Xiaosong Li
Joshua J. Goings, Hang Hu, Chao Yang, and Xiaosong Li. Reinforcement learning Configura- tion Interaction.Journal of Chemical Theory and Computation, 17(9):5482–5491, sep 2021
2021
-
[14]
Williams-Young, Norm M
David B. Williams-Young, Norm M. Tubman, Carlos Mejuto-Zaera, and Wibe A. de Jong. A parallel, distributed memory implementation of the adaptive sampling Configuration Interac- tion method.The Journal of Chemical Physics, 158(21):214109, June 2023
2023
-
[15]
Kammeraad, and Paul M
Duy-Khoi Dang, Joshua A. Kammeraad, and Paul M. Zimmerman. Advances in parallel heat bath Configuration Interaction.The Journal of Physical Chemistry A, 127(1):400–411, January 2023
2023
-
[16]
Holmes, Sandeep Sharma, and C
Junhao Li, Matthew Otten, Adam A. Holmes, Sandeep Sharma, and C. J. Umrigar. Fast semistochastic heat-bath Configuration Interaction.The Journal of Chemical Physics, 149(21):214110, December 2018
2018
-
[17]
Yann Garniron, Thomas Applencourt, Kevin Gasperich, Anouar Benali, Anthony Fert´ e, Julien Paquier, Barth´ el´ emy Pradines, Roland Assaraf, Peter Reinhardt, Julien Toulouse, Pierrette Barbaresco, Nicolas Renon, Gr´ egoire David, Jean-Paul Malrieu, Micka¨ el V´ eril, Michel Caffarel, Pierre-Fran¸ cois Loos, Emmanuel Giner, and Anthony Scemama. Quantum Pac...
-
[18]
E. Y. Loh, J. E. Gubernatis, R. T. Scalettar, Steven R. White, D. J. Scalapino, and R. L. Sugar. Sign problem in the numerical simulation of many-electron systems.Physical Review B, 41:9301–9307, 1990
1990
-
[19]
Booth, Alex J
George H. Booth, Alex J. W. Thom, and Ali Alavi. Fermion Monte Carlo without fixed nodes: A game of life, death, and annihilation in Slater determinant space.The Journal of Chemical Physics, 131(5):054106, 2009
2009
-
[20]
J. S. Spencer, N. S. Blunt, and W. M. C. Foulkes. The sign problem and population dynamics in the full Configuration Interaction quantum Monte Carlo method.The Journal of Chemical Physics, 136:054110, 2012
2012
-
[21]
Booth, and Ali Alavi
Deidre Cleland, George H. Booth, and Ali Alavi. Communications: Survival of the fittest: Accelerating convergence in full Configuration-Interaction quantum Monte Carlo.The Journal of Chemical Physics, 132(4):041103, 2010
2010
-
[22]
F. R. Petruzielo, A. A. Holmes, H. J. Changlani, M. P. Nightingale, and C. J. Umrigar. Semistochastic projector Monte Carlo method.Physical Review Letters, 109:230201, 2012. 29
2012
-
[23]
N. S. Blunt, J. J. Shepherd, D. K. K. Lee, G. H. Booth, and A. Alavi. Semi-stochastic full Configuration Interaction quantum Monte Carlo: Developments and application.The Journal of Chemical Physics, 142:184107, 2015
2015
-
[24]
Booth, Andreas Gr¨ uneis, Georg Kresse, and Ali Alavi
George H. Booth, Andreas Gr¨ uneis, Georg Kresse, and Ali Alavi. Towards an exact description of electronic wavefunctions in real solids.Nature, 493(7432):365–370, January 2013
2013
-
[25]
Anderson, Nick S
Kai Guther, Robert J. Anderson, Nick S. Blunt, Nikolay A. Bogdanov, Deidre Cleland, Nike Dattani, Werner Dobrautz, Khaldoon Ghanem, Peter Jeszenszki, Niklas Liebermann, Gio- vanni Li Manni, Alexander Y. Lozovoi, Hongjun Luo, Dongxia Ma, Florian Merz, Catherine Overy, Markus Rampp, Pradipta Kumar Samanta, Lauretta R. Schwarz, James J. Shepherd, Simon D. Sm...
2020
-
[26]
Steven R. White. Density-matrix algorithms for quantum renormalization groups.Physical Review B, 48:10345–10356, 1993
1993
-
[27]
White and Richard L
Steven R. White and Richard L. Martin. Ab initio quantum chemistry using the Density Matrix Renormalization Group.The Journal of Chemical Physics, 110:4127–4130, 1999
1999
-
[28]
The Density-Matrix Renormalization Group in the age of matrix product states.Annals of Physics, 326(1):96–192, January 2011
Ulrich Schollw¨ ock. The Density-Matrix Renormalization Group in the age of matrix product states.Annals of Physics, 326(1):96–192, January 2011
2011
-
[29]
Spin-adapted Density Matrix Renormalization Group algorithms for quantum chemistry.The Journal of Chemical Physics, 136(12):124121, March 2012
Sandeep Sharma and Garnet Kin-Lic Chan. Spin-adapted Density Matrix Renormalization Group algorithms for quantum chemistry.The Journal of Chemical Physics, 136(12):124121, March 2012
2012
-
[30]
Low communication high performanceab ini- tioDensity Matrix Renormalization Group algorithms.The Journal of Chemical Physics, 154(22):224116, June 2021
Huanchen Zhai and Garnet Kin-Lic Chan. Low communication high performanceab ini- tioDensity Matrix Renormalization Group algorithms.The Journal of Chemical Physics, 154(22):224116, June 2021
2021
-
[31]
Distributed-memory DMRG via sparse and dense parallel tensor contractions
Ryan Levy, Edgar Solomonik, and Bryan K Clark. Distributed-memory DMRG via sparse and dense parallel tensor contractions. November 2020
2020
-
[32]
Xantheas, Martin Ganahl, and ¨Ors Legeza
Andor Menczer, Maarten van Damme, Alan Rask, Lee Huntington, Jeff Hammond, Sotiris S. Xantheas, Martin Ganahl, and ¨Ors Legeza. Parallel implementation of the Density Matrix Renormalization Group method achieving a quarter petaFLOPS performance on a single DGX- H100 GPU node.Journal of Chemical Theory and Computation, 20(19):8397–8404, October
-
[33]
Publisher: American Chemical Society
-
[34]
Enhua Xu, Motoyuki Uejima, and Seiichiro L. Ten-no. Towards near-exact solutions of molec- ular electronic structure: Full Coupled-Cluster Reduction with a second-order perturbative correction.The Journal of Physical Chemistry Letters, 11(22):9775–9780, November 2020. Publisher: American Chemical Society
2020
-
[35]
Eriksen and J¨ urgen Gauss
Janus J. Eriksen and J¨ urgen Gauss. Many-body expanded full Configuration Interaction. I. Weakly correlated regime.Journal of Chemical Theory and Computation, 14(10):5180–5191, October 2018. Publisher: American Chemical Society
2018
-
[36]
Eriksen and J¨ urgen Gauss
Janus J. Eriksen and J¨ urgen Gauss. Many-body expanded full Configuration Interaction. II. Strongly correlated regime.Journal of Chemical Theory and Computation, 15(9):4873–4884, September 2019. Publisher: American Chemical Society. 30
2019
-
[37]
Coordinate descent full Configuration Interaction
Zhe Wang, Yingzhou Li, and Jianfeng Lu. Coordinate descent full Configuration Interaction. Journal of Chemical Theory and Computation, 15(6):3558–3569, 2019
2019
-
[38]
Coordinatewise descent methods for leading eigen- value problem.SIAM Journal on Scientific Computing, 41(4):A2681–A2716, 2019
Yingzhou Li, Jianfeng Lu, and Zhe Wang. Coordinatewise descent methods for leading eigen- value problem.SIAM Journal on Scientific Computing, 41(4):A2681–A2716, 2019
2019
-
[39]
Coordinate descent full Configura- tion Interaction for excited states.Journal of Chemical Theory and Computation, 19(21):7731– 7739, 2023
Zhe Wang, Zhiyuan Zhang, Jianfeng Lu, and Yingzhou Li. Coordinate descent full Configura- tion Interaction for excited states.Journal of Chemical Theory and Computation, 19(21):7731– 7739, 2023
2023
-
[40]
Parallel multicoordinate descent methods for full Configuration Interaction.Journal of Chemical Theory and Computation, 21(5):2325–2337, 2025
Yuejia Zhang, Weiguo Gao, and Yingzhou Li. Parallel multicoordinate descent methods for full Configuration Interaction.Journal of Chemical Theory and Computation, 21(5):2325–2337, 2025
2025
-
[41]
Yingzhou Li and Jianfeng Lu. Optimal orbital selection for full Configuration Interaction (OptOrbFCI): Pursuing the basis set limit under a budget.Journal of Chemical Theory and Computation, 16(10):6207–6221, 2020
2020
-
[42]
Bjørn O. Roos. The complete active space self-consistent field method and its applications in electronic structure calculations. In K. P. Lawley, editor,Advances in Chemical Physics: Ab Initio Methods in Quantum Chemistry Part 2, volume 69, pages 399–445. John Wiley & Sons, New York, 1987
1987
-
[43]
Triangularized orthogonalization-free method for solving extreme eigenvalue problems.Journal of Scientific Computing, 93(3):63, 2022
Weiguo Gao, Yingzhou Li, and Bichen Lu. Triangularized orthogonalization-free method for solving extreme eigenvalue problems.Journal of Scientific Computing, 93(3):63, 2022
2022
-
[44]
Stephen J. Wright. Coordinate descent algorithms.Mathematical Programming, 151:3–34, 2015
2015
-
[45]
Eigen v3.http://eigen.tuxfamily.org, 2010
Ga¨ el Guennebaud, Benoˆ ıt Jacob, et al. Eigen v3.http://eigen.tuxfamily.org, 2010
2010
-
[46]
Robin hood hashing.https://github.com/martinus/ robin-hood-hashing
Martin Leitner-Ankerl. Robin hood hashing.https://github.com/martinus/ robin-hood-hashing. Accessed: 2025-10-08
2025
-
[47]
Andersen, and Michael Kaminsky
Bin Fan, David G. Andersen, and Michael Kaminsky. MemC3: Compact and concurrent memcache with dumber caching and smarter hashing. InProceedings of the 10th USENIX Symposium on Networked Systems Design and Implementation (NSDI 13), pages 371–384. USENIX Association, 2013
2013
-
[48]
Andersen, Michael Kaminsky, and Michael J
Xi Wang Li, David G. Andersen, Michael Kaminsky, and Michael J. Freedman. Algorith- mic improvements for fast concurrent cuckoo hashing. InProceedings of the Ninth European Conference on Computer Systems (EuroSys ’14), pages 27:1–27:14. ACM, 2014
2014
-
[49]
A high-performance concurrent hash table (libcuckoo).https:// github.com/efficient/libcuckoo
Efficient Systems Lab. A high-performance concurrent hash table (libcuckoo).https:// github.com/efficient/libcuckoo. Accessed: 2025-10-08
2025
-
[50]
Eriksen, Tyler A
Janus J. Eriksen, Tyler A. Anderson, J. Emiliano Deustua, Khaldoon Ghanem, Diptarka Hait, Mark R. Hoffmann, Seunghoon Lee, Daniel S. Levine, Ilias Magoulas, Jun Shen, Norm M. Tubman, K. Birgitta Whaley, Enhua Xu, Yuan Yao, Ning Zhang, Ali Alavi, Garnet Kin-Lic Chan, Martin Head-Gordon, Wenjian Liu, Piotr Piecuch, Sandeep Sharma, Seiichiro L. Ten- no, C. J...
2020
-
[51]
QUESTDB: A database of highly accurate ex- citation energies for the electronic structure community.WIREs Computational Molecular Science, 11(5):e1517, 2021
Micka¨ el V´ eril, Anthony Scemama, Michel Caffarel, Filippo Lipparini, Mario Boggio-Pasqua, Denis Jacquemin, and Pierre-Fran¸ cois Loos. QUESTDB: A database of highly accurate ex- citation energies for the electronic structure community.WIREs Computational Molecular Science, 11(5):e1517, 2021
2021
-
[52]
A mountaineering strategy to excited states: Highly accurate reference energies and benchmarks.Journal of Chemical Theory and Computation, 14(8):4360–4379, 2018
Pierre-Fran¸ cois Loos, Anthony Scemama, Aymeric Blondel, Yann Garniron, Michel Caffarel, and Denis Jacquemin. A mountaineering strategy to excited states: Highly accurate reference energies and benchmarks.Journal of Chemical Theory and Computation, 14(8):4360–4379, 2018
2018
-
[53]
Quest database of highly accurate excitation energies
Pierre-Fran¸ cois Loos and collaborators. Quest database of highly accurate excitation energies. https://github.com/pfloos/QUESTDB, 2021. Accessed: 2025-10-08
2021
-
[54]
Correlated electrons in high-temperature superconductors.Reviews of Modern Physics, 66(3):763–840, 1994
Elbio Dagotto. Correlated electrons in high-temperature superconductors.Reviews of Modern Physics, 66(3):763–840, 1994
1994
-
[55]
Turney, Andrew C
Justin M. Turney, Andrew C. Simmonett, Robert M. Parrish, Edward G. Hohenstein, Francesco A. Evangelista, Justin T. Fermann, Benjamin J. Mintz, Lori A. Burns, Jeremiah J. Wilke, Micah L. Abrams, Nicholas J. Russ, Matthew L. Leininger, Curtis L. Janssen, Ed- ward T. Seidl, Wesley D. Allen, Henry F. Schaefer, Rollin A. King, Edward F. Valeev, C. David Sherr...
2012
-
[56]
R. W. D. Nickalls. A new approach to solving the cubic: Cardan’s solution revealed.The Mathematical Gazette, 77(480):354–359, 1993. A Determining Stepsize in Problem(15)and(28) We describe in detail how to minimize the quartic polynomial in Problem (15) and (28), thereby determining the optimal step sizeη. In Problem (15), we have h(η) =f(c (ℓ) +ηe i(ℓ+1)...
1993
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.