Recognition: 2 theorem links
· Lean TheoremSPADE: Faster Drug Discovery by Learning from Sparse Data
Pith reviewed 2026-05-08 17:54 UTC · model grok-4.3
The pith
SPADE finds 10 high-quality ligands for a new protein target with an average of 40 tests.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
SPADE introduces a novel approach to ligand selection that requires only 40 tests on average to find 10 high-quality ligands. In one-vs-one comparisons, SPADE outperforms deep learning and Bayesian optimization methods on more proteins, achieving median improvements of 7%-32% in sample efficiency. SPADE is also 10x faster than its closest competitor at scoring candidate drugs.
What carries the argument
SPADE, an iterative ligand selection algorithm that updates its choices after each round of sparse test results to prioritize high-quality binders.
If this is right
- Early screening for novel protein targets can reach a usable set of binders after far fewer experiments.
- Computational ranking of large candidate libraries becomes practical because scoring runs much faster.
- Methods that depend on large amounts of pre-existing protein data are no longer required for initial rounds of discovery.
- The same selection logic can be reused across different proteins without retraining on protein-specific datasets.
Where Pith is reading between the lines
- The sparse-learning idea could be combined with docking simulations to further cut the number of physical tests needed.
- Similar selection rules might reduce trial costs in other experimental domains where each measurement is expensive.
- Success in real pipelines would still require showing that ligands labeled high-quality by the method advance through later drug-development stages at higher rates than random selection.
Load-bearing premise
The reported gains on the evaluated proteins and ligand sets will hold when the method is applied to entirely new proteins with no existing measurements.
What would settle it
Apply SPADE and the competing methods to a previously untested protein target, run each until 10 ligands pass independent binding assays, and compare the exact number of tests used by each.
Figures

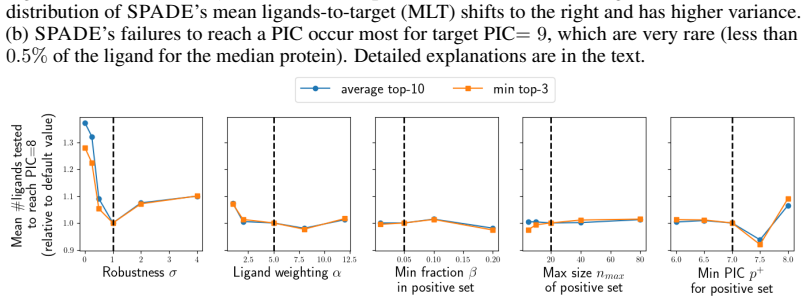

read the original abstract
Drug discovery seeks molecules (ligands) that bind strongly and selectively to a target protein. However, fewer than 5% of candidate ligands pass the bar for even the early stages of drug discovery. Furthermore, we want methods that work for novel proteins for which we have no prior data. Starting from scratch, we have to iteratively select and test candidate ligands such that we find enough ligands of the desired quality in as few tests as possible. Our proposed algorithm, named SPADE, introduces a novel approach to ligand selection that requires only 40 tests on average to find 10 high-quality ligands. In one-vs-one comparisons, SPADE outperforms deep learning and Bayesian optimization methods on more proteins, achieving median improvements of 7%-32% in sample efficiency. SPADE is also 10x faster than its closest competitor at scoring candidate drugs. Dataset and code is available at https://anonymous.4open.science/r/SPADE_Fast_Drug_Discovery_by_Learning_from_Sparse_Data-F028/README.md
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript introduces SPADE, a novel algorithm for ligand selection in drug discovery that learns from sparse data on novel proteins with no prior information. It claims that SPADE requires only 40 tests on average to identify 10 high-quality ligands, outperforms deep learning and Bayesian optimization baselines in one-vs-one comparisons on more proteins (with median sample-efficiency gains of 7-32%), and runs 10x faster than its closest competitor when scoring candidates. Dataset and code are released via an anonymous repository link.
Significance. If the performance claims are supported by rigorous, reproducible experiments with clear protocols, the work could meaningfully advance sample-efficient active learning for molecular design in drug discovery. The emphasis on generalization to truly novel targets and computational speed addresses practical bottlenecks in early-stage screening.
major comments (3)
- [Abstract] Abstract: The central performance claims (average 40 tests for 10 ligands, 7-32% median improvements, 10x scoring speedup) are presented without any reference to the number of proteins evaluated, the specific datasets or oracles used, the train/test protein splits, statistical tests, or baseline implementation details. This information is load-bearing for assessing whether the method truly generalizes to novel proteins with zero prior data.
- [Evaluation section] Evaluation section (presumably §4 or §5): The one-vs-one comparisons and reported median gains require explicit documentation of protein selection criteria (to rule out scaffold/family leakage), the precise definition of 'high-quality ligands' (e.g., affinity cutoff or other threshold), and how the oracle realism aligns with downstream therapeutic value. Absent these, the sample-efficiency claims cannot be verified.
- [Method section] Method section: The novel ligand-selection mechanism in SPADE must be accompanied by ablations or complexity analysis that isolates its contribution to the reported speed and efficiency gains relative to the deep learning and Bayesian optimization baselines.
minor comments (2)
- [Abstract] Abstract: 'Dataset and code is available' should read 'Datasets and code are available'.
- [Abstract / Data availability] The anonymous repository link should be replaced with a permanent, non-anonymous URL or a detailed description of the released assets to support reproducibility.
Simulated Author's Rebuttal
We thank the referee for their constructive and detailed comments on our manuscript. We address each major comment below in a point-by-point manner and indicate the revisions we will implement to strengthen the paper.
read point-by-point responses
-
Referee: [Abstract] Abstract: The central performance claims (average 40 tests for 10 ligands, 7-32% median improvements, 10x scoring speedup) are presented without any reference to the number of proteins evaluated, the specific datasets or oracles used, the train/test protein splits, statistical tests, or baseline implementation details. This information is load-bearing for assessing whether the method truly generalizes to novel proteins with zero prior data.
Authors: We agree that the abstract would be improved by including brief references to the experimental context supporting the claims. In the revised manuscript, we will add a concise clause noting the number of proteins evaluated, the datasets and oracles employed, the zero-prior train/test splits for novel proteins, the statistical tests used, and that baselines follow standard implementations from the literature. This will be done while respecting abstract length constraints by focusing on the most essential details and directing readers to the main text for full protocols. revision: yes
-
Referee: [Evaluation section] Evaluation section (presumably §4 or §5): The one-vs-one comparisons and reported median gains require explicit documentation of protein selection criteria (to rule out scaffold/family leakage), the precise definition of 'high-quality ligands' (e.g., affinity cutoff or other threshold), and how the oracle realism aligns with downstream therapeutic value. Absent these, the sample-efficiency claims cannot be verified.
Authors: We acknowledge that these details should be stated more explicitly and prominently. The manuscript already covers protein selection from diverse families with dissimilarity thresholds to avoid leakage, defines high-quality ligands via affinity cutoffs and ranking within the oracle, and uses oracles based on validated docking and experimental data. To address the comment directly, we will insert a dedicated paragraph at the beginning of the Evaluation section that consolidates these criteria, adds a summary table of datasets and splits, and includes a short discussion of oracle limitations relative to full therapeutic validation. This will make the claims fully verifiable without changing any results. revision: yes
-
Referee: [Method section] Method section: The novel ligand-selection mechanism in SPADE must be accompanied by ablations or complexity analysis that isolates its contribution to the reported speed and efficiency gains relative to the deep learning and Bayesian optimization baselines.
Authors: We agree that isolating the novel sparse adaptation component is important for crediting the observed gains. The current method section describes the mechanism and includes some runtime analysis, but we will expand it with a new subsection containing targeted ablations (SPADE with and without the sparse module versus the baselines) and a detailed complexity breakdown showing how the embedding-based scoring yields the reported speedup. Key ablation results and tables will be moved from the supplement into the main text to directly address this point. revision: yes
Circularity Check
No circularity: empirical performance claims rest on external benchmarks and code release, not self-referential definitions or fitted inputs.
full rationale
The abstract and available description present SPADE as an iterative ligand-selection algorithm whose central claims are measured average test counts (40 for 10 ligands) and median improvements (7-32%) versus deep learning and Bayesian optimization baselines on specific proteins. These are reported experimental outcomes on held-out datasets rather than quantities derived by construction from the method's own parameters or prior self-citations. No equations, uniqueness theorems, or ansatzes are invoked in the provided text that reduce the reported efficiencies to fitted inputs renamed as predictions. The evaluation setup, while subject to generalization questions, is independent of the algorithm's internal logic and is supported by released code, satisfying the criteria for a non-circular finding.
Axiom & Free-Parameter Ledger
Lean theorems connected to this paper
-
IndisputableMonolith/Cost (Jcost = ½(x+x⁻¹)−1)washburn_uniqueness_aczel unclear?
unclearRelation between the paper passage and the cited Recognition theorem.
ℓ(C(x), y) := max{0, 1 − y · C(x)} ... E_{x∼N(x_i, σ²I)}[ℓ(C(x), y=1)] = s_i · Φ(s_i/(σ‖w‖)) + σ‖w‖ · φ(s_i/(σ‖w‖))
-
IndisputableMonolith/Foundation (parameter-free forcing chain)reality_from_one_distinction unclear?
unclearRelation between the paper passage and the cited Recognition theorem.
score(x_j) := Σ_{i∈S+} α^{p_i} · C_i(x_j), with α=5, σ=1, β=0.05, n_max=20, p+=7
What do these tags mean?
- matches
- The paper's claim is directly supported by a theorem in the formal canon.
- supports
- The theorem supports part of the paper's argument, but the paper may add assumptions or extra steps.
- extends
- The paper goes beyond the formal theorem; the theorem is a base layer rather than the whole result.
- uses
- The paper appears to rely on the theorem as machinery.
- contradicts
- The paper's claim conflicts with a theorem or certificate in the canon.
- unclear
- Pith found a possible connection, but the passage is too broad, indirect, or ambiguous to say the theorem truly supports the claim.
Reference graph
Works this paper leans on
-
[1]
Robust High-Dimensional Classification From Few Positive Examples
Deepayan Chakrabarti and Benjamin Fauber. Robust High-Dimensional Classification From Few Positive Examples. InProceedings of the Thirty-First International Joint Conference on Artificial Intelligence, pages 1952–1958, Vienna, Austria, July 2022. International Joint Conferences on Artificial Intelligence Organization. ISBN 978-1-956792-00-3. doi: 10.24963...
1952
-
[2]
Improving the Generalizability of Protein-Ligand Binding Predictions with AI-Bind.Nat
Ayan Chatterjee, Robin Walters, Zohair Shafi, Omair Shafi Ahmed, Michael Sebek, Deisy Morselli Gysi, Rose Yu, Tina Eliassi-Rad, Albert-László Barabási, and Giulia Menichetti. Improving the Generalizability of Protein-Ligand Binding Predictions with AI-Bind.Nat. Commun., 14:1989, 2023
1989
-
[3]
arXiv preprint arXiv:2010.09885 (2020)
Seyone Chithrananda, Gabriel Grand, and Bharath Ramsundar. ChemBERTa: large-scale self-supervised pretraining for molecular property prediction.arXiv preprint arXiv:2010.09885, 2020
-
[4]
Davis, Jeremy P Hunt, Sanna Herrgård, Pietro Ciceri, Lisa M
Mindy I. Davis, Jeremy P Hunt, Sanna Herrgård, Pietro Ciceri, Lisa M. Wodicka, Gabriel Pallares, Michael Hocker, Daniel K. Treiber, and Patrick P. Zarrinkar. Comprehensive analysis of kinase inhibitor selectivity.Nature Biotechnology, 29:1046–1051, 2011. URL https: //api.semanticscholar.org/CorpusID:32070305
2011
-
[5]
Joseph L. Durant, Burton A. Leland, Douglas R. Henry, and James G. Nourse. Reoptimization of MDL Keys for Use in Drug Discovery.Journal of Chemical Information and Computer Sciences, 42(6):1273–1280, November 2002. ISSN 0095-2338. doi: 10.1021/ci010132r. URL https://doi.org/10.1021/ci010132r. Publisher: American Chemical Society
-
[6]
Ben Fauber. Accurate Prediction of Ligand-Protein Interaction Affinities with Fine-Tuned Small Language Models.ArXiv, abs/2407.00111v1, 2024
-
[7]
Genome Scale Enzyme–Metabolite and Drug–Target Interaction Predictions Using the Signature Molecular Descriptor.Bioinformatics, 24(2):225–233, 2007
Jean-Loup Faulon, Milind Misra, Shawn Martin, Ken Sale, and Rajat Sapra. Genome Scale Enzyme–Metabolite and Drug–Target Interaction Predictions Using the Signature Molecular Descriptor.Bioinformatics, 24(2):225–233, 2007. ISSN 1367-4803
2007
-
[8]
Gilson, Tiqing Liu, Michael Baitaluk, George Nicola, Linda Hwang, and Jenny Chong
Michael K. Gilson, Tiqing Liu, Michael Baitaluk, George Nicola, Linda Hwang, and Jenny Chong. Bindingdb in 2015: A public database for medicinal chemistry, computational chemistry and systems pharmacology.Nucleic Acids Research, 44:D1045 – D1053, 2015. URL https: //api.semanticscholar.org/CorpusID:8843610
2015
-
[9]
Weiße, and Antonia S
Rohan Gorantla, Alžbeta Kubincová, Andrea Y . Weiße, and Antonia S. J. S. Mey. From Proteins to Ligands: Decoding Deep Learning Methods for Binding Affinity Prediction.J. Chem. Inf. Model., 64(7):2496–2507, 2024
2024
-
[10]
TABM: Advancing Tabular Deep Learning With Parameter-Efficient Ensembling
Yury Gorishniy, Akim Kotelnikov, and Artem Babenko. TABM: Advancing Tabular Deep Learning With Parameter-Efficient Ensembling. 2025
2025
-
[11]
GAUCHE: A library for Gaussian processes in chemistry.Advances in Neural Information Processing Systems, 36, 2024
Ryan-Rhys Griffiths, Leo Klarner, Henry Moss, Aditya Ravuri, Sang Truong, Yuanqi Du, Samuel Stanton, Gary Tom, Bojana Rankovic, Arian Jamasb, et al. GAUCHE: A library for Gaussian processes in chemistry.Advances in Neural Information Processing Systems, 36, 2024
2024
-
[12]
Accurate predictions on small data with a tabular foundation model.Nature, 637(8045):319–326, 2025
Noah Hollmann, Samuel Müller, Lennart Purucker, Arjun Krishnakumar, Max Körfer, Shi Bin Hoo, Robin Tibor Schirrmeister, and Frank Hutter. Accurate predictions on small data with a tabular foundation model.Nature, 01 2025. doi: 10.1038/s41586-024-08328-6. URL https://www.nature.com/articles/s41586-024-08328-6
-
[13]
DeepPur- pose: A Deep Learning Library for Drug–Target Interaction Prediction.Bioinformatics, 36: 5545 – 5547, 2020
Kexin Huang, Tianfan Fu, Lucas Glass, Marinka Zitnik, Cao Xiao, and Jimeng Sun. DeepPur- pose: A Deep Learning Library for Drug–Target Interaction Prediction.Bioinformatics, 36: 5545 – 5547, 2020
2020
-
[14]
MolTrans: Molecular Interaction Transformer for Drug–Target Interaction Prediction.Bioinformatics, 37:830 – 836, 2020
Kexin Huang, Cao Xiao, Lucas Glass, and Jimeng Sun. MolTrans: Molecular Interaction Transformer for Drug–Target Interaction Prediction.Bioinformatics, 37:830 – 836, 2020. 10
2020
-
[15]
TransDTI: Transformer-Based Language Models for Estimating DTIs and Building a Drug Recommendation Workflow.ACS Omega, 7: 2706 – 2717, 2022
Yogesh Kalakoti, Shashank Yadav, and Durai Sundar. TransDTI: Transformer-Based Language Models for Estimating DTIs and Building a Drug Recommendation Workflow.ACS Omega, 7: 2706 – 2717, 2022
2022
-
[16]
Joshua D Kangas, Armaghan W Naik, and Robert F Murphy. Efficient discovery of responses of proteins to compounds using active learning.BMC Bioinformatics, 15(1), December 2014. ISSN 1471-2105. doi: 10.1186/1471-2105-15-143. URL https://bmcbioinformatics. biomedcentral.com/articles/10.1186/1471-2105-15-143 . Publisher: Springer Sci- ence and Business Media LLC
-
[17]
Kimber, Yonghui Chen, and Andrea V olkamer
Talia B. Kimber, Yonghui Chen, and Andrea V olkamer. Deep Learning in Virtual Screening: Recent Applications and Developments.Int. J. Mol. Sci., 22:4435, 2021
2021
-
[18]
Target Selection in Drug Discovery.Nat
Jonathan Knowles and Gianni Gromo. Target Selection in Drug Discovery.Nat. Rev. Drug Discov., 2:63–69, 2003
2003
-
[19]
DeepConv-DTI: Prediction of Drug-Target Interactions via Deep Learning with Convolution on Protein sequences.PLOS Comput
Ingoo Lee, Jongsoo Keum, and Hojung Nam. DeepConv-DTI: Prediction of Drug-Target Interactions via Deep Learning with Convolution on Protein sequences.PLOS Comput. Biol., 15(6):e1007129, 06 2019
2019
-
[20]
Lenselink, Niels ten Dijke, Brandon Bongers, George Papadatos, Herman W
Eelke B. Lenselink, Niels ten Dijke, Brandon Bongers, George Papadatos, Herman W. T. van Vlijmen, Wojtek Kowalczyk, Adriaan P. IJzerman, and Gerard J. P. van Westen. Beyond the Hype: Deep Neural Networks Outperform Established Methods Using a ChEMBL Bioactivity Benchmark Set.J. Cheminform., 9:45, 2017
2017
-
[21]
MONN: A Multi-objective Neural Network for Predicting Compound-Protein Interactions and Affinities
Shuya Li, Fangping Wan, Hantao Shu, Tao Jiang, Dan Zhao, and Jianyang Zeng. MONN: A Multi-objective Neural Network for Predicting Compound-Protein Interactions and Affinities. Cell Syst., pages 308–322.e11, 2020
2020
-
[22]
Lombardino and John A
Joseph G. Lombardino and John A. Lowe III. The Role of the Medicinal Chemist in Drug Discovery — Then and Now.Nat. Rev. Drug Discov., 3:853–862, 2004
2004
-
[23]
Martin, Prasenjit Mukherjee, David C
Eric J. Martin, Prasenjit Mukherjee, David C. Sullivan, and Johanna M. Jansen. Profile-QSAR: A Novel meta-QSAR Method that Combines Activities across the Kinase Family To Accurately Predict Affinity, Selectivity, and Cellular Activity.J. Chem. Inf. Model., 51(8):1942–1956, 2011
1942
-
[24]
Martin, Valery R
Eric J. Martin, Valery R. Polyakov, Xiang-Wei Zhu, Li Tian, Prasenjit Mukherjee, and Xin Liu. All-Assay-Max2 pQSAR: Activity Predictions as Accurate as Four-Concentration IC50s for 8558 Novartis Assays.J. Chem. Inf. Model., 59(10):4450–4459, 2019
2019
-
[25]
Steijaert, Jörg Kurt Wegner, Hugo Ceulemans, Djork-Arné Clevert, and Sepp Hochreiter
Andreas Mayr, Günter Klambauer, Thomas Unterthiner, Marvin N. Steijaert, Jörg Kurt Wegner, Hugo Ceulemans, Djork-Arné Clevert, and Sepp Hochreiter. Large-Scale Comparison of Machine Learning Methods for Drug Target Prediction on ChEMBL.Chem. Sci., 9:5441–5451, 2018
2018
-
[26]
Directory of useful decoys, enhanced (DUD-E): better ligands and decoys for better benchmarking.Journal of medicinal chemistry, 55(14):6582–6594, 2012
Michael M Mysinger, Michael Carchia, John J Irwin, and Brian K Shoichet. Directory of useful decoys, enhanced (DUD-E): better ligands and decoys for better benchmarking.Journal of medicinal chemistry, 55(14):6582–6594, 2012
2012
-
[27]
Oliveira, Rita C Guedes, and Andre O Falcao
Pedro F. Oliveira, Rita C Guedes, and Andre O Falcao. Inferring Molecular Inhibition Potency with AlphaFold Predicted Structures.Sci. Rep., 14:8252, 2024
2024
-
[28]
WideDTA: Prediction of Drug- Target Binding Affinity.ArXiv, abs/1902.04166, 2019
Hakime Öztürk, Elif Ozkirimli Olmez, and Arzucan Özgür. WideDTA: Prediction of Drug- Target Binding Affinity.ArXiv, abs/1902.04166, 2019
-
[29]
IJzerman, Andreas Bender, and Florian Nigsch
Shardul Paricharak, Adriaan P. IJzerman, Andreas Bender, and Florian Nigsch. Analysis of Iterative Screening with Stepwise Compound Selection Based on Novartis In-house HTS Data. ACS Chemical Biology, 11(5):1255–1264, May 2016. ISSN 1554-8929, 1554-8937. doi: 10.1021/acschembio.6b00029. URL https://pubs.acs.org/doi/10.1021/acschembio. 6b00029. 11
-
[30]
URL https://www.biorxiv.org/content/early/2025/06/18/2025.06.14.659707
Saro Passaro, Gabriele Corso, Jeremy Wohlwend, Mateo Reveiz, Stephan Thaler, Vignesh Ram Somnath, Noah Getz, Tally Portnoi, Julien Roy, Hannes Stark, David Kwabi-Addo, Dominique Beaini, Tommi Jaakkola, and Regina Barzilay. Boltz-2: Towards accurate and efficient binding affinity prediction.bioRxiv, 2025. doi: 10.1101/2025.06.14.659707
-
[31]
Plowright, Craig Johnstone, Jan Kihlberg, Jonas Pettersson, Graeme Robb, and Richard A
Alleyn T. Plowright, Craig Johnstone, Jan Kihlberg, Jonas Pettersson, Graeme Robb, and Richard A. Thompson. Hypothesis Driven Drug Design: Improving Quality and Effectiveness of the Design-Make-Test-Analyse Cycle.Drug Discov. Today, 17(1):56–62, 2012
2012
-
[32]
Mateusz Praski, Jakub Adamczyk, and Wojciech Czech. Benchmarking pretrained molecular embedding models for molecular representation learning.arXiv preprint arXiv:2508.06199, 2025
-
[33]
Lawrence Zitnick, Jerry Ma, and Rob Fergus
Alexander Rives, Joshua Meier, Tom Sercu, Siddharth Goyal, Zeming Lin, Jason Liu, Demi Guo, Myle Ott, C. Lawrence Zitnick, Jerry Ma, and Rob Fergus. Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences.PNAS, 2019. doi: 10.1101/622803. URLhttps://www.biorxiv.org/content/10.1101/622803v4
-
[34]
Extended-connectivity fingerprints
David Rogers and Mathew Hahn. Extended-Connectivity Fingerprints.Journal of Chemical Information and Modeling, 50(5):742–754, May 2010. ISSN 1549-9596. doi: 10.1021/ ci100050t. URL https://doi.org/10.1021/ci100050t. Publisher: American Chemical Society
-
[35]
Ross, Chao Lu, Guido Scarabelli, Steven K
Gregory A. Ross, Chao Lu, Guido Scarabelli, Steven K. Albanese, Evelyne Houang, Robert Abel, Edward D Harder, and Lingle Wang. The Maximal and Current Accuracy of Rigorous Protein-Ligand Binding Free Energy Calculations.Commun. Chem., 6:222, 2023
2023
-
[36]
Sadybekov, Anastasiia V
Arman A. Sadybekov, Anastasiia V . Sadybekov, Yongfeng Liu, Christos Iliopoulos-Tsoutsouvas, Xi-Ping Huang, Julie E. Pickett, Blake Houser, Nilkanth Patel, Ngan K. Tran, Fei Tong, Nikolai Zvonok, M. K. Jain, Olena V . Savych, Dmytro S. Radchenko, Spyros P. Nikas, Nicos A. Petasis, Yurii S. Moroz, Bryan L. Roth, Alexandros Makriyannis, and Vsevolod Katritc...
2021
-
[37]
Christina E. M. Schindler, Hannah Baumann, Andreas Blum, Dietrich Böse, Hans-Peter Buch- staller, Lars Burgdorf, Daniel Cappel, Eugene Chekler, Paul Czodrowski, Dieter Dorsch, Merveille K. I. Eguida, Bruce Follows, Thomas Fuchß, Ulrich Grädler, Jakub Gunera, Theresa Johnson, Lebrun Catherine Jorand, Srinivasa Karra, Markus Klein, Tim Knehans, Lisa Koetzne...
2020
-
[38]
Reed M. Stein, Ying Yang, Trent E. Balius, Matt J. O’Meara, Jiankun Lyu, Jennifer Young, Khanh Tang, Brian K. Shoichet, and John J. Irwin. Property-Unmatched Decoys in Docking Benchmarks.Journal of Chemical Information and Modeling, 61(2):699–714, February 2021. ISSN 1549-9596, 1549-960X. doi: 10.1021/acs.jcim.0c00598. URL https://pubs.acs. org/doi/10.102...
-
[39]
HyperPCM: Robust Task-Conditioned Modeling of Drug–Target Interactions.J
Emma Svensson, Pieter-Jan Hoedt, Sepp Hochreiter, and Günter Klambauer. HyperPCM: Robust Task-Conditioned Modeling of Drug–Target Interactions.J. Chem. Inf. Model., 64:2539 – 2553, 2024
2024
-
[40]
Swinney and Jason Anthony
David C. Swinney and Jason Anthony. How Were New Medicines Discovered?Nat. Rev. Drug Discov., 10:507–519, 2011
2011
-
[41]
Thafar, Mona Alshahrani, Somayah Albaradei, Takashi Gojobori, Magbubah Essack, and Xin Gao
Maha A. Thafar, Mona Alshahrani, Somayah Albaradei, Takashi Gojobori, Magbubah Essack, and Xin Gao. Affinity2Vec: Drug-Target Binding Affinity Prediction Through Representation Learning, Graph Mining, and Machine Learning.Sci. Rep., 12:4751, 2022
2022
-
[42]
LIT-PCBA: an unbiased data set for machine learning and virtual screening.Journal of chemical information and modeling, 60(9):4263–4273, 2020
Viet-Khoa Tran-Nguyen, Célien Jacquemard, and Didier Rognan. LIT-PCBA: an unbiased data set for machine learning and virtual screening.Journal of chemical information and modeling, 60(9):4263–4273, 2020. 12
2020
-
[43]
Dahlgren, Jeremy R
Lingle Wang, Yujie Wu, Yuqing Deng, Byungchan Kim, Levi Pierce, Goran Krilov, Dmitry Lupyan, Shaughnessy Robinson, Markus K. Dahlgren, Jeremy R. Greenwood, Donna Lee Romero, Craig E. Masse, Jennifer L. Knight, Thomas Steinbrecher, Thijs Beuming, Wolfgang Damm, Edward D Harder, Woody Sherman, Mark L. Brewer, Ron Wester, Mark A. Murcko, Leah L. Frye, Ramy F...
2015
-
[44]
Waring, John Edmund Arrowsmith, Andrew R
Michael J. Waring, John Edmund Arrowsmith, Andrew R. Leach, Paul D. Leeson, Sam Mandrell, Robert M. Owen, Garry Pairaudeau, William D. Pennie, Stephen D. Pickett, Jibo Wang, Owen Wallace, and Alexander Weir. An Analysis of the Attrition of Drug Candidates from Four Major Pharmaceutical Companies.Nat. Rev. Drug Discov., 14:475–486, 2015
2015
-
[45]
Deep-Learning-Based Drug-Target Interaction Prediction.J
Ming Wen, Zhimin Zhang, Shaoyu Niu, Haozhi Sha, Rui Yang, Yong-Huan Yun, and Hongmei Lu. Deep-Learning-Based Drug-Target Interaction Prediction.J. Proteome Res., 16:1401–1409, 2017
2017
-
[46]
Whitehead, Benedict W J Irwin, Peter A
Thomas M. Whitehead, Benedict W J Irwin, Peter A. Hunt, Matthew D. Segall, and Gareth John Conduit. Imputation of Assay Bioactivity Data Using Deep Learning.J. Chem. Inf. Model., 59: 1197–1204, 2019
2019
-
[47]
Prediction of Drug–Target Interaction Networks from the Integration of Chemical and Genomic Spaces.Bioinformatics, 24:i232 – i240, 2008
Yoshihiro Yamanishi, Michihiro Araki, Alex Gutteridge, Wataru Honda, and Minoru Kanehisa. Prediction of Drug–Target Interaction Networks from the Integration of Chemical and Genomic Spaces.Bioinformatics, 24:i232 – i240, 2008. 13 A Proofs Proof of Theorem 3.1.The proof is similar to Theorem 1 of [1]. We have Ex∼N(x i,σ2I) [ℓ(C(x), y= 1)] =Ex∼N(x i,σ2I) ma...
2008
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.