Recognition: unknown
Feature Dimensionality Outweighs Model Complexity in Breast Cancer Subtype Classification Using TCGA-BRCA Gene Expression Data
Pith reviewed 2026-05-08 12:27 UTC · model grok-4.3
The pith
Feature dimensionality outweighs model complexity in classifying breast cancer subtypes from gene expression data, with logistic regression performing most stably.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
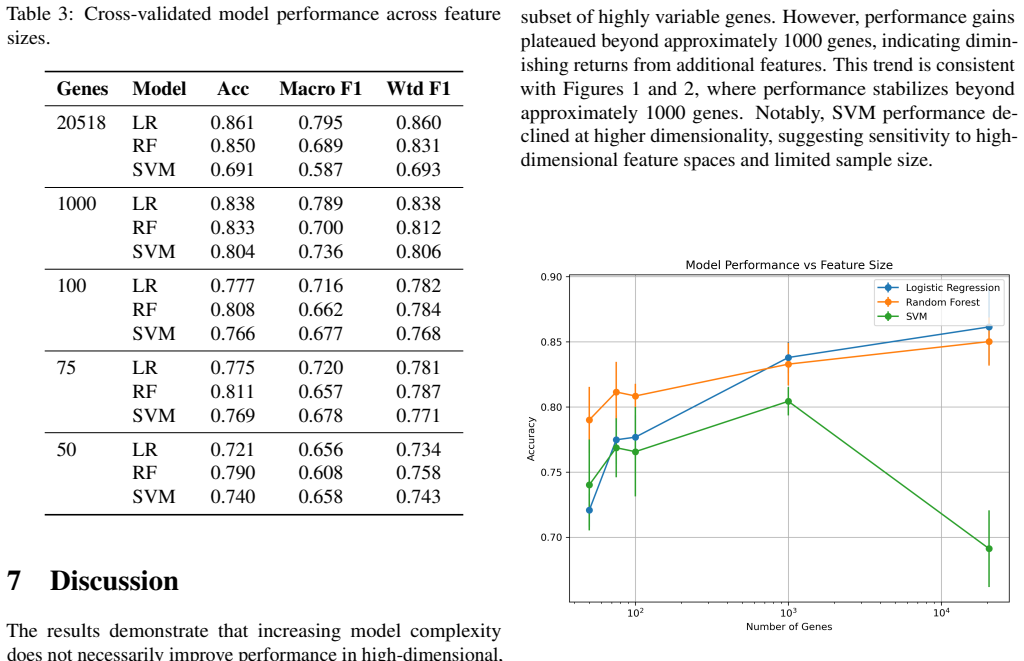
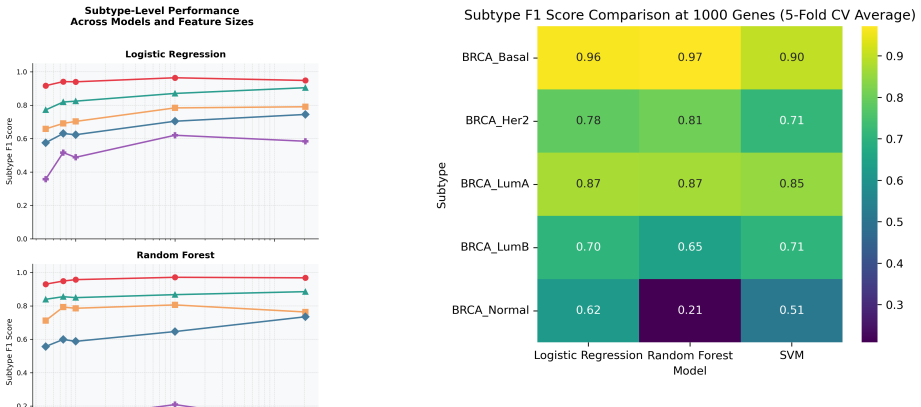
The authors demonstrate that when classifying breast cancer subtypes using TCGA-BRCA gene expression profiles, the dimensionality of the feature set has a greater influence on performance than the intrinsic complexity of the model. Logistic regression exhibited the most consistent results across all subtypes using macro F1 scores, improving detection of minority classes, whereas random forest struggled with rare subtypes despite high overall accuracy, and SVM performance varied notably with the number of genes selected.
What carries the argument
The systematic comparison of logistic regression, random forest, and SVM classifiers trained on incrementally larger sets of highly variable genes, evaluated via stratified cross-validation with both accuracy and macro-averaged F1 score.
If this is right
- Logistic regression provides more reliable detection of all breast cancer subtypes, including rare ones, compared to complex models.
- Macro F1 score reveals performance issues hidden by accuracy metrics in imbalanced classification tasks.
- Feature selection and dimensionality are key to effective modeling in high-dimensional genomic data.
- Simpler linear models may suffice or excel in biological classification problems with limited samples.
Where Pith is reading between the lines
- Similar patterns may hold in other high-dimensional imbalanced genomic classification tasks, favoring simpler models for stability.
- The interpretability of logistic regression could make it preferable for clinical applications beyond raw performance.
- Testing these models on independent breast cancer cohorts would help confirm if the dimensionality effect generalizes beyond TCGA-BRCA.
Load-bearing premise
The selection of highly variable genes introduces no systematic bias related to subtypes, and macro F1 differences arise from model behavior rather than unstated preprocessing or tuning variations.
What would settle it
Repeating the analysis with all genes included or using an alternative feature selection method like differential expression analysis and checking if the relative performance of logistic regression on macro F1 remains superior.
Figures


read the original abstract
Accurate classification of breast cancer subtypes from gene expression data is critical for diagnosis and treatment selection. However, such datasets are characterized by high dimensionality and limited sample size, posing challenges for machine learning models. In this study, we evaluate the impact of model complexity and feature selection on subtype classification performance using TCGA-BRCA gene expression data. Logistic regression, random forest, and support vector machine (SVM) models were trained using varying numbers of highly variable genes (50 to 20,518). Performance was evaluated using stratified 5-fold cross-validation and assessed with accuracy and macro F1 score. While all models achieved high accuracy, macro F1 analysis revealed substantial differences in subtype-level performance. Logistic regression demonstrated the most stable and balanced performance across subtypes, including improved detection of rare classes. Random forest underperformed on minority subtypes despite strong overall accuracy, while SVM showed sensitivity to feature dimensionality. These findings highlight the importance of model simplicity, evaluation metrics, and feature selection in high-dimensional biological classification tasks.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The paper claims that feature dimensionality (number of highly variable genes from 50 to 20,518) outweighs model complexity in breast cancer subtype classification on TCGA-BRCA gene expression data. Logistic regression, random forest, and SVM are compared via stratified 5-fold cross-validation using accuracy and macro F1, concluding that logistic regression yields the most stable and balanced subtype-level performance while highlighting the importance of model simplicity and metrics in high-dimensional settings.
Significance. If the central empirical findings hold after correcting for potential preprocessing artifacts, the work would usefully illustrate the pitfalls of accuracy-only evaluation in imbalanced genomic classification and the value of simpler linear models when sample sizes are small relative to dimensionality, providing practical guidance for similar TCGA-style analyses.
major comments (1)
- [Abstract/Methods] Abstract and methods description of the experimental pipeline: highly variable gene selection is not stated to occur inside each cross-validation fold. Selecting HVGs on the full matrix before stratified splitting risks data leakage whose severity increases with dimensionality; this directly threatens the load-bearing claim that performance gaps (especially logistic regression stability on rare subtypes) reflect intrinsic dimensionality-vs-complexity effects rather than selection bias.
minor comments (2)
- No information is provided on hyperparameter tuning procedures, regularization strengths, or random seeds, making it difficult to reproduce the reported macro F1 values.
- The manuscript does not report per-fold variance, confidence intervals, or statistical significance tests comparing models or dimensionality settings.
Simulated Author's Rebuttal
We thank the referee for their careful reading and for identifying a key point of methodological clarity. We address the major comment below and will revise the manuscript accordingly to strengthen the description of our pipeline.
read point-by-point responses
-
Referee: [Abstract/Methods] Abstract and methods description of the experimental pipeline: highly variable gene selection is not stated to occur inside each cross-validation fold. Selecting HVGs on the full matrix before stratified splitting risks data leakage whose severity increases with dimensionality; this directly threatens the load-bearing claim that performance gaps (especially logistic regression stability on rare subtypes) reflect intrinsic dimensionality-vs-complexity effects rather than selection bias.
Authors: We agree that the abstract and methods sections did not explicitly state the location of HVG selection within the cross-validation procedure, and we thank the referee for catching this omission. In the actual implementation, HVG selection (via the same variance-based criterion) was performed independently on the training portion of each stratified fold before model fitting, with the selected genes then applied to the held-out test fold. This was done to avoid leakage. We will revise the methods section (and update the abstract if space permits) to describe the full pipeline step-by-step, explicitly noting that feature selection occurs inside the CV loop. These changes will eliminate ambiguity and directly support the validity of our dimensionality-vs-complexity comparisons. revision: yes
Circularity Check
No circularity: purely empirical ML evaluation with no derivations or self-referential claims
full rationale
The paper reports experimental results from training logistic regression, random forest, and SVM on TCGA-BRCA gene expression data using varying numbers of highly variable genes, evaluated via stratified 5-fold CV with accuracy and macro F1. No equations, first-principles derivations, or predictions are present. The central claim that feature dimensionality outweighs model complexity is a direct summary of observed performance differences, not a reduction to fitted inputs or self-citations by construction. No self-citation load-bearing, ansatz smuggling, or renaming of known results occurs. The study is self-contained against external benchmarks (public TCGA data and standard ML metrics), so the derivation chain has no circular steps.
Axiom & Free-Parameter Ledger
free parameters (1)
- Number of highly variable genes
Reference graph
Works this paper leans on
-
[1]
Random forests.Machine Learning, 45(1):5–32, 2001
Leo Breiman. Random forests.Machine Learning, 45(1):5–32, 2001
2001
-
[2]
Bhupender Singh Chhikara et al. Deep learning based model for breast cancer subtype classification.arXiv preprint arXiv:2111.03923, 2021
-
[3]
Support-vector networks
Corinna Cortes and Vladimir Vapnik. Support-vector networks. Machine Learning, 20(3):273–297, 1995
1995
-
[4]
A guide to deep learning in healthcare
Andre Esteva et al. A guide to deep learning in healthcare. Nature Medicine, 25:24–29, 2019
2019
-
[5]
Golub, Donna K
Todd R. Golub, Donna K. Slonim, Pablo Tamayo, Christine Huard, Michelle Gaasenbeek, Jill P. Mesirov, Hilary Coller, Mignon L. Loh, James R. Downing, Mark A. Caligiuri, Clara D. Bloomfield, and Eric S. Lander. Molecular classification of can- cer: class discovery and class prediction by gene expression monitoring.Science, 286(5439):531–537, 1999
1999
-
[6]
An introduction to variable and feature selection.Journal of Machine Learning Research, 3:1157–1182, 2003
Isabelle Guyon and Andre Elisseeff. An introduction to variable and feature selection.Journal of Machine Learning Research, 3:1157–1182, 2003
2003
-
[7]
Haibo He and Edwardo A. Garcia. Learning from imbalanced data.IEEE Transactions on Knowledge and Data Engineering, 21(9):1263–1284, 2009
2009
-
[8]
Libbrecht and William S
Maxwell W. Libbrecht and William S. Noble. Machine learning applications in genetics and genomics.Nature Reviews Genetics, 16(6):321–332, 2015
2015
-
[9]
Super- vised risk predictor of breast cancer based on intrinsic subtypes
Joel S Parker, Michael Mullins, Maggie CU Cheang, et al. Super- vised risk predictor of breast cancer based on intrinsic subtypes. Journal of Clinical Oncology, 27(8):1160–1167, 2009
2009
-
[10]
A review of feature selection techniques in bioinformatics.Bioinformatics, 23(19):2507–2517, 2007
Yvan Saeys, I ˜naki Inza, and Pedro Larra ˜naga. A review of feature selection techniques in bioinformatics.Bioinformatics, 23(19):2507–2517, 2007. 7
2007
-
[11]
Sprefuged: A method to select a predictive func- tion for a gene expression data based feature selection workflow
Hector Sanz, Clarissa Valim, Esteban Vegas, Josep M Oller, and Ferran Reverter. Sprefuged: A method to select a predictive func- tion for a gene expression data based feature selection workflow. PLOS ONE, 12(12), 2017
2017
-
[12]
A systematic analysis of performance measures for classification tasks.Information Processing & Management, 45(4):427–437, 2009
Marina Sokolova and Guy Lapalme. A systematic analysis of performance measures for classification tasks.Information Processing & Management, 45(4):427–437, 2009
2009
-
[13]
Gene expression patterns of breast carcinomas distinguish tumor sub- classes with clinical implications.Proceedings of the National Academy of Sciences, 98(19):10869–10874, 2001
Therese Sorlie, Charles M Perou, Robert Tibshirani, et al. Gene expression patterns of breast carcinomas distinguish tumor sub- classes with clinical implications.Proceedings of the National Academy of Sciences, 98(19):10869–10874, 2001
2001
-
[14]
Aliferis, Ioannis Tsamardi- nos, Douglas Hardin, and Shawn Levy
Alexander Statnikov, Constantin F. Aliferis, Ioannis Tsamardi- nos, Douglas Hardin, and Shawn Levy. A comprehensive evalua- tion of multicategory classification methods for microarray gene expression cancer diagnosis.Bioinformatics, 21(5):631–643, 2005
2005
-
[15]
Weinstein et al
John N. Weinstein et al. The cancer genome atlas pan-cancer analysis project.Nature Genetics, 45(10):1113–1120, 2013
2013
-
[16]
Breast cancer type classifica- tion using machine learning.Journal of Personalized Medicine, 11(2):61, 2021
Jiande Wu and Chindo Hicks. Breast cancer type classifica- tion using machine learning.Journal of Personalized Medicine, 11(2):61, 2021
2021
-
[17]
Rna- seq-based breast cancer subtypes classification using machine learning approaches.Computational and Mathematical Methods in Medicine, 2020:4737969, 2020
Zhezhou Yu, Zhuo Wang, Xiangchun Yu, and Zhe Zhang. Rna- seq-based breast cancer subtypes classification using machine learning approaches.Computational and Mathematical Methods in Medicine, 2020:4737969, 2020. 8
2020
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.